|
The
current name is PRIMARY CNS
LYMPHOMAS (PCNSL).
Few other tumors have had so many different names, which include
malignant lymphoma, non Hodgkin’s lymphoma, microgliomatosis,
microglioma, reticuluim cell sarcoma, microgrliomatosis, perivascular
sarcoma, reticulendothelial sarcoma, malignant reticulosis, malignant,
reticulo-endotheliosis, histocytic lymphoma, immunoblastic sarcoma and
granulomatous encephalitism. Much of the older American literature has
referred to these tumors as reticulum cell sarcomas because of the
similarity with tumors of lymph node origin. In Europe the term
microglioma was preferred because of the microlglial like silver staining
properties of cells within these tumors. It now appears however that these
cells are reactive and not an intrinsic component of the tumor.
The
above list of names reflects the past uncertainty regarding the
cell of origin of this tumor. These tumors were, now, found to be almost
exclusively B cell lymphomas of the non Hodgkin’s type, as confirmed by immunocytochemical and
latterly molecular genetic studies.
Epidemiology:
PCNSL
represents rare form of non-Hodgkin's lymphoma, and account for <1% of
all primary brain tumors and 1% of non-Hodgkin’s lymphomas. However
the incidence is increasing both in immunosuppressed and in non
immunosupporessed patients and this important findings is unexplained.
Although
diagnosed predominantly among immunocompetent, those immunosuppressed
patients are at particular risk of developing PCNSL and they can be
divided into four groups:
Patients
with AIDS of whom approximately 3% will develop PCNSL and in whom PCNSL
is the most common cause of a non-infectious intracranial mass lesion.
Reports
suggest that there is annual incidence of primary cerebral lymphoma
between 0.22%-1.6% in renal and cardiac transplant recepients. This
represents an increased risk to that of the general population by 350
fold.
Patients
with congenital immunodeficiency states where the risk of developing
PCNSL is approximately 4%.
Other
rare associations of the tumor with drug or disease induced
immunosuppresion include sarcoidosis, systemic lupus erthematous,
Sjogren’s syndrome, vasculitis, rheumatoid arthritis, idiopathic
thrombocytopenic purpura and progressive multifocal leucoencephalopathy.
The
associated conditions without evidence of immunosuppression include
tuberculosis, multiple sclerosis and other malignancies.
Patient
of any age can be affected, although most non-immunosuppressed patients
present in their 5th-7th decades with a mean age of
57. Most series of non-immunosuppressed patients show an increased
incidence in men with an approximate male to female ratio of
2:1. Patients with AIDS presenting with the disease are usually male
and age 30-40.
Pathology:
Although
the tumor's origin principally as a B cell lymphoma has been determined,
how these neoplastic cells come to proliferate within the central nervous
system is not understood and why they are so frequently multifocal at
presentation is primary CNS tumor that may completely disappear with
steroids adds to the interesting nature of the tumor.
The CNS has neither lymphatic circulation nor physiological
accumulations of lymphoid tissue: a fact which has led to different
theories regarding the origin of the neoplastic lymphoid cells in the
CNS.
There are two possible theories of origin.
Firstly, a non neoplastic reactive population of lymphocytes is
attracted into the CNS by an inflammatory or infectious event and
then transformed locally into a neoplastic clone. The transforming
factor has been proposed to be a virus. This clone may then express
a binding molecule specific to the CNS or CNS vascular endothelium,
allowing the cells to spread via the bloodstream but only adhering to the
CNS. One piece of supporting evidence for this theory is the
knowledge of chronic viral infections of the CNS which evade immune
surveillance.
The second theory suggests that a clone of B lymphocytes possessing
a CNS specific binding site proliferate outside the CNS and are
transformed into neoplastic cells which circulate but only bind to the
CNS. The site of origin remains obscure while the clone
proliferates within the CNS. This latter theory incorporates a
proposed vascular spread with a CNS binding site, which explains the high
incidence of multifocality at presentation, and also the peculiar
attraction for the CNS. However it does not account for the increased
frequency of occurrence adjacent to the ventricle or within the frontal
lobes. A blood borne tumor, even if possessing a specific CNS
marker, should occur at the more common sites for metastatic tumor i.e.
the parietal lobe while an intrinsic tumor would be expected to occur
most often in the largest area of brain, i.e. the frontal lobe.
It
is likely that while the etiology of PCNSL in the non-immunosuppressed
remains obscure, in immunosuppressed individuals with reduced immune
surveillance (under T cell control) the Epstein Barr virus (EBV) may be
directly implicated. The EBV genome has been identified in cells
from PCNSL in both immunosuppressed and non-immunosuppressed
patients. However in one report, using an in-situ hybridization
technique with a biotinylated DNA probe to the internal repeat regions of
the EBV genome, evidence of EBV was found in all 5 cases of
immunosuppressed PCNSL but in only 2 of 43 non-immunosuppressed patients
(Geddes, 1992). The association of EBV genome to PCNSL in
immunosuppressed patients could represent secondary infection; however
the EBV genome was identified in neoplastic cells rather than adjacent
reactive cells.
In
vitro studies of peripheral lymphoma cell lines suggest that the
expression of activated c-myc (a proto-oncogene) in combination with
infection by EBV causes tumorigenesis, however, studies of PCNSL have
usually failed to show these c-myc rearrangements.
Further use of molecular
biological techniques may identify specific CNS markers, other viral
DNA or consistent patterns of genetic abnormalities associated with these
tumors and thereby lead to a better understanding of their origin.
Primary CNS
lymphoma usually manifests in the brain (30-50%), leptomeninges (10-25%),
eye (10-20%), or spinal cord.
PCNSLs
may occur in any location in the brain; but they are most commonly found
in the cerebral hemispheres.
The
frontal lobe is the most common site and the occipital lobe the least
common.
They
usually lie deeply within the basal ganglia or adjacent to the
ventricle.
Spread
may involve the corpus callosum, producing the so called ‘butterfly
tumor’ or be subependymal or meningeal.
Lesions
below the tentorium occur and when present are most often situated in the
cerebellum.
Multiple
lesions
are found at presentation in 30-50%, but are more common in the
immunosuppressed at 50-80%. Other primary site include the eyes, cranial
or spinal nerves and although spinal meningeal disease occurs it is much more
common with recurrence and neuroaxis dissemination.
Macroscopically the lesions can
vary greatly in appearance, some being well defined yellow-white lesions
quite distinct from normal white matter, other may be grey or
brown. Some may show diffuse discoloration and swelling or appear
grossly normal. Cyst formation is rare but necrosis and hemorrhage may be
present. The tumors may act as space occupying lesions with
significant edema and mass effect or they may be ill defined and diffuse. Typically
they replace rather than displace normal brain with resultant little mass
effect.
Microscopically PCNSL are
richly cellular with large round or oval cells with a lymphoid
appearance. They have round, oval or kidney shaped nuclei.
Necrosis is common; mitotic figures and varying degrees of pleomorphism
may also be present. One characteristic feature of these tumors is
their perivascular orientation with infiltration and distension of the
perivascular spaces. This is demonstrated by reticulin stains which
show concentric rings around blood vessels representing reduplication of
reticulin. Endoepthelial proliferation is not typical but spread via
perivascular spaces in adjacent brain is, resulting in a diffuse
infiltration with neoplastic cells being found distant to the macroscopic
borders of the tumor.
Leptomeningeal spread is also
characteristic. They may resemble bacterial meningitis with semi liquid
tissue lying with the meninges or producing thickening of the cranial or
spinal nerves. This tissue may fill the sulci and basal cisterns
and obscure the cortical surface. In the spinal cord the cauda
equina may be matted together. The cells do not form follicles or
nodules. Immunological and molecular genetic studies have shown that
these are B cell lymphomas. T cell lymphomas occur very rarely in
only 1-2% although T cells may be present when they are likely to
represent an inflammatory response.
Systemic
spread of PCNSL is found at necropsy in 7-34%; however in the majority of
cases this is clinically silent and usually occurs in the presence of
recurrent CNS disease.
Secondary
involvement of the CNS by systemic lymphoma will develop in
2-10% of cases of non Hogkin’s lymphoma. This usually present as
extradural compression in the spinal canal but other sites, including
intracranial extradural, meningeal and intraparenchymal deposits, do
occur, although rarely. Systemic Hodgkin’s disease seldom involves
the CNS and primary CNS Hodgkin’s lymphoma is extremely rare.
Clinical
presentation:
The
signs and symptoms
produced by PCNSL reflect their location and pathological behavior.
The
presenting history is often only a few months and most patients present
with a space occupying lesion or lesions producing local deficits.
As a frontal location is commonest, accordingly motor dysfunction,
personality or other neuro psychological deficits appear frequently in
20-30%. Subtle changes of personality, depression or memory loss
may also reflect diffuse involvement of white matter tracts, particularly
in association with periventricular and corpus callosum lesion.
Patients
with mass lesions and raised intracranial pressure may present with
headache but this may also suggest meningeal involvement and other signs
of meningism or cranial and spinal nerve dysfunction should be looked
for.
Seizures
occur in 5-25% of patients, The pattern of clinical presentation
may mimic other pathology such as cerebrovascular accident or
encephalitis. Infratentorial lesions cause ataxia, cranial nerve lesions
or other disturbances of brain stem function.
Spinal
deposits occur frequently with recurrence but primary intramedullary non
Hodgkin’s lymphoma is very rare.
There
is a significant incidence of visual disturbance and this may be either
lymphoma affecting the uvea, choroid or retina or pathology affecting the
intracranial visual pathways.
Slit
lamp examination of patients with suspected intracranial disease should
be carried out as it may show occult disease.
Radiology:
|
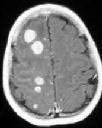
The
CT and MRI findings confirm that PCNSL are usually seen as a frontal
and /or periventricular mass but with minimal mass effect.
On
unenhanced CT these lesions are iso or hyperdense, consistent
with their increased cellularity. Contrast enhancement is usually
marked and uniform with a variable amount of abnormal density
surrounding the enhancing lesion. This may be true edema, but may
also indicate tumor invasion. Less common radiological
appearances include lesions with significant mass effect and ring
enhancement.
The
ring enhancement has been reported more often in cases arising
in the immunosuppressed.
Non-enhancing
lesions have also been reported.
|
|
|
|
|
Lymphomatosis-MRI
|
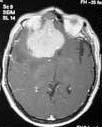
PCNSL-MRI
|
|
MRI
shows
the tumors to be iso or hyperintense to grey matter on T2 weighted images
with homogeneous enhancement after contrast. Multiplicity of
lesions is found in 40% of MRIs performed but MRI has not been shown to
significatly better than CT in making the diagnosis.
In
one study using CT 29% of patients had multiple lesions and in this
series the prognosis of this group was not significantly worse than in
those who had single lesions on CT.
Positron
emission tomography (PET) with HC-methyl-L-methionine has been
shown to be useful in following the response to therapy as these lesions
may temporarily disappear on CT after steroids or other therapy.
This
rapid disappearance is unique among cerebral tumors but multiple
sclerosis and sarcoidosis are other possibilities.
The
differential diagnosis of a mass lesion includes metastases high
grade astrocytoma, secondary lymphoma, meningioma, abcess and
toxoplasmosis. Radiological features such as a periventricular site
or mass with ependymal/meningeal enhancement are highly suggestive and
may increase the chance of a positve diagnosis being obtained from the
CSF.
Diagnosis:
Although
the diagnosis may be suspected radiologically, histological verification
is required.
In
10% of patients, particularly those with subependymal and periventricular
involvement, lumbar puncture and CSF cytology may provide the
diagnosis. Immunostaining
with B and T cell markers is very useful in the identification and
classification of PCNSL and may demostrate monoclonal populations even if
the cells appear cytologically benign.
The
CSF biochemistry commonly shows a raised protein level with normal glucose
content, and white blood cell pleocytosis.
If
CSF cannot be obtained safely or cytology is inconclusive, needle biopsy
should be performed preferably using CT guided stereotaxy. If
craniotomy is performed and per-operative smear indicates lymphoma then
aggressive surgical excision is not justified. The extent of
surgical excision has not been shown to have an effect on quality or
length of survival.
The
surgeon should also bear in mind that these tumors usually show a very
good early response to adjuvant therapy even in patients with a
significant tumor bulk.
Although
staging is carried out in many centers and as part of some trial
protocols, systemic non Hodgkin’s lymphoma is found in <5% patients
presenting with cerebral lymphoma. There are no reported cases of occult
systemic lymphoma presenting as a CNS lymphoma. Thus CT scanning of
thorax and abdomen and bone marrow examination are unnecessary unless
clinically indicated. However, if it can be performed safely, CSF
should be obtained as positive cytology suggests ependymal or meningeal
involvement and the patient should then be considered for intrathecal
methotrexate therapy.
Slit
lamp examination of the eye and serology for syphilis and HIV antigen
should also be carried out.
Treatment:
The
neurosurgeon will see these tumors with increasing frequency over time
but the role of surgery in management is principally to suspect the
diagnosis and then confirm it by biopsy.
After
histological confirmation the oncologist/radiotherapist will contribute
most to overall therapy.
Steroids have an
immediate and dramatic impact on symptoms. There are a number of
reports of tumors disappearing on cT after steroids. This is a direct
cytotoxic effect but, while in some remission may be sustained for months,
most relapse within weeks. Disappearance of the tumor after
initiation of steroid therapy will make sterotactic biopsy impossible
although the tumor may still be detected by PET scanning. Some recommend
withholding steroid therapy until after biopsy unless the patient is at
risk. The reappearance of the tumor either with or without cessation of
steroids will allow biopsy if CSF is negative.
Radiotherapy: Traditional
therapy for primary cerebral lymphoma has been post operative
radiotherapy. These tumors are generally radiotherapy responsive,
although less so than when they arise outside the CNS. However this
response is not sustained and 80-95% of cases recur, usually between
10-12 months after therapy. The prognosis with radiotherapy alone
after biopsy is a median survival of 10-18 months although much worse
results have been reported. Radiotherapy is usually given to the brain
down to C2 and if there is ocular involvement the orbits are
included. A total dose of 4000 to 5000 cGy is recommended to the
whole brain but whether there is any additional benefit from a boost to
the tumor itself is unclear although recommended.
Most
PCNSL recur within the fields of radiation treatment. PCNSL exhibit
a high rate of multicentricity and diffusely infiltrate the brain
parenchyma, deposits in so called retinal and subarchnoid sanctuaries
also occur and these may not be demonstrated on enhanced CT or MRI. In
light of these observations and the poor prognosis despite radiotherapy,
chemotherapy is being increasingly used.
Chemotherapy: Most modern chemotherapeutic
regimes, of which there are several, include high dose MTX which
penetrates the CNS relatively well but may cause leukoencephlopathy when
used following radiotherapy.
There
are a number of different chemotherapy regimes in use for pre or post
irradiation treatment and a combination of pre and post irradiation
chemotherapy has also been employed. Results with chemotherapy have
improved median survival from 14 months to 18 to 44 months.
Deferring the use of radiotherapy after chemotherapy until recurrence has
also been advocated by some. Treatment of patients with radiological or
cytological evidence of meningeal disease is recommended with
intrathercal MTX either via a lumbar puncture or a ventricular catheter
connected to an Ommaya reservoir.
DeAngelis has employed a
combined modality therapy with systemic MTX and intrathecal MTX via an
Ommaya reservoir into the ventricle pre-radiotherapy, followed by
cytarabine systemically post radiotherapy.
Neuwelt
have
been using an intensive regime including blood brain barrier modulation
with intracarotid or vertebral mannitol prior to intra-arterial
chemotherapy. Both groups have published improved survival data
with a median survival around 43 months.
Whether
or not these intensive treatment regimes will prove generally applicable
or effective and what the patient selection criteria should be for
different therapies remains to be clarified.
Nevertheless
there is accumulating evidence to suggest that patients should be offered
chemotherapy following biopsy and prior to radiotherapy.
Patients
with AIDs presenting with PCNSL are usually treated with steroids and
radiotherapy as their already immunosuppressed condition often rules out
chemotherapy. The mean survival time for this group is 3-5.5
months.
Prognosis:
Approximately
80-95% if tumors recur locally within the radiation fields. 93% of
recurrences are confined to the CNS with neuroaxis dissemination in 60%.
Systemic spread of PCNSL is found at necropsy in 7-34%; however in the
majority of cases this is clinically silent and rarely occurs in the
absence of recurrent CNS disease. The 5 year survival is approximately
2-5% and most patients die from local disease within 2 years of diagnosis
with a median survival of 10-18 months after radiotherapy.
With
chemotherapy with or without radiotherapy median survival is improved to
17-44 months.
Positive
prognositc indicators include a solitary intracranial lesion, favorable
histology and the administration of radiotherapy or chemotherapy.
Adverse
factors include periventricular/meningeal lesions and immunosuppression.
It has been suggested that an elevated CSF protein >0.6g per
litre is the strongest negative prognostic indicator while the other
negative factors included performance status and age over 60.
|
