|
OLIGODENDROGLIOMA:
Oligodendrocytes are specialized
neuraglia that form myelin in the CNS, first recognized by Robertson in
1900. In 1926, Bailey and Cushing first
described oligodendrogliomas in a histogenic classification of gliomas.
They are a subgroup of
gliomas with distinctive morphological characteristics.
At present, oligodendrogliomas are
thought to constitute approximately 5% to 10% of all primary tumors of the brain, but by using expanded
criteria, they may well represent up to one third. In fact, morphologic
criteria are vague and highly subjective and the histologic diagnosis,
therefore, remains highly controversial and unsatisfactory. It has been
suggested that the traditional 'diffuse fibrillary astrocytoma' are in
fact made of isolated neoplastic oligodendrocytes which are entrapped in
a fibrillary background composed of axons and fibrillary reactive
gliosis.
They are most often
diagnosed in patients who are middle aged, though a
bimodal distribution has been described with a second, smaller peak among children and
adolescents.
It
has been demonstrated that oligodendroglial neoplasms usually have loss
of 1p and 19q whereas astrocytomas of the progressive type frequently
contain mutations of the TP53 gene, and that 9p loss and CDKN2A deletions
are associated with progression from well-differentiated to anaplastic
oligodendrogliomas.
|
Oligodendrogliomas most commonly involve the cerebral hemisphere.
Involvement of the frontal and temporal lobes is particularly
common.
Rare cerebellar
oligodendroliomas have been described.
Patients usually present with a
history of seizures.
CT scanning,
typically, reveals a supratentorial lesion with nodular calcifications
and surrounding edema. Contrast enhancement is seen in 60% of cases. On
MRI, they are hypo to isodense on T1 and hyperdense on T2 images
with heterogenous enhancement.
Oligodendrogliomas generally
expand the cortex diffusely giving it a hypertrophic appearance, while
the underlying white matter may show cysts of varying sizes. While
they appear grey and friable, calcified granules can often be felt when
cut with the knife.
However, in the
oligodendrogliomas seen in India, the calcification has been found to
be
minimal in contrast to that
seen in similar tumors in the west.
|
|
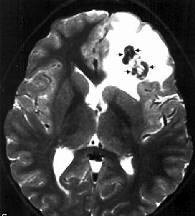
|
|
Oligodendroglioma with
calcification-MRI T2
|
|
|
Histologically, it consists
of a uniform pattern of cells containing round to oval nuclei
surrounded by a clear cytoplasmic rim
(fried egg appearance). In
addition, a characteristic 'chicken wire' vascular pattern can be seen
to demarcate discrete groups of tumor cells. Necrosis, atypia, and
mitosis can be seen in anaplastic variants.
Oligodendrogliomas are probably
derived from multipotent cells; this would explain the fact many
oligodendrogliomas are of
mixed glial composition (mixed
gliomas-oligoastrocytoma).
According to Rubinstein almost
half the oligodendrogliomas consist of mixed cell forms. The
combination is mainly with an astrocytoma grade II or III. This is not
surprising in view of the close association of astrocytes and
oligodendrocytes in most parts of the normal brain. In one study, 15
per cent were found to be mixed gliomas. In 11%, there was a
combination of astrocytoma and oligodendroglioma, in two of
oligodendroglioma and ependymoma and in two of ependymoma and
astrocytoma.
|
|
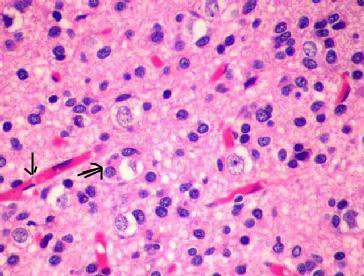
|
|
Oligodendroglioma (H&E) -
the
artefactual perinuclear satellitosis(double arrow) and
delicate thin walled blood vessels(arrow) which
produces
the
fried egg appearance.
|
|
Grading of these tumors
is difficult as most of the oligodendrogliomas show monomorphism as
regards the oligodendroglial tumor proper, but frequently they are
associated with an astrocytoma grade II.
Anaplastic
oligodendrogliomas (AO) are uncommon. They have the features of high
grade neoplasm e.g. increased mitotic activity, necrosis, vascular
endothelial proliferation and hypercellularity. Their natural history,
prognosis, and optimal management are not yet fully understood. These
tumors still maintain the rounded shapes of their nuclei and the arciform
vasculature. These tumors can progress sometimes to Glioblastoma multiforme
and, in the process, incorporate many astrocytes. However, they are
associated with a better prognosis and response to multimodality therapy
based on specific molecular changes.
Management include, aggressive excision and
post operative radiation.
Chemotherapy with PCV (procarbazine,
CCNU, and vincristine) has also been advocated, particularly in mixed and
anaplastic variants. Oligodendrogliomas have
been the focus of considerable interest over the past decade, ever since
they were recognized as chemosensitive tumors. A recent report
suggests that alterations of chromosome arms 1p and 19q are associated
with chemotherapeutic response and overall survival in anaplastic
oligodendroglioma patients treated with procarbazine, lomustine, and
vincristine chemotherapy.
While postoperative survival is
generally longer with oligodendrolioma, its prognosis is ‘notoriously
uncertain’. The morbidity and mortality profile
for oligodendrogliomas is much better than for astrocytic tumors.
However, it also depends on tumor location and pressure effects, as with
any other intracranial lesion.
5 year survival rates for patients with
oligodendrogliomas are commonly in the range of 50% to 65%. Early diagnosis,
complete excision, and low grade histology predict a better prognosis.
Glial tumors of unknown origin:
They include Gliomatosis Cerebri, Astroblastomas, and Choroid gliomas.
1) Gliomatosis Cerebri
(diffuse cerebral gliomatosis):
The term
"gliomatosis cerebri" is used to describe a diffusely proliferative glial neoplasm
involving large regions of the
brain, characterized by an infiltrative nature rather than by the
formation of a distinct tumor mass.
They are rare.
Although found anywhere throughout, the CNS, multilobar hemispheric
involvement is most common.
Usually, the
involvement is diffuse.
Symptoms at
presentation are frequently those of seizure or
headache. Most patients present between third and fourth decades.
The duration of
symptoms may be several months to many years.
CT may reveal
subtle hypo to isodense changes with diffuse mass effects. MRI may be
more illustrative.
Occasionally,
there may be minimal contrast hetereogenous enhancement.
Multiple sclerosis,
encephalitis, and leukodystrophies must be ruled out. CSF cytology
is unremarkable.
Positron emission
tomography (PET) scans using 11C-L- methionine show isotope
accumulation in the diffusely infiltrative tumorous area with greater
accuracy than CT or MRI.
Many are diagnosed
at autopsy.
Grossly, there is
diffuse enlargement. The gyri may be slightly enlarged with normal
architecture.
Biopsy is required
for diagnosis.
Histologically, a
diffuse infiltration of astrocytic cells bearing a spindle like
pattern is seen;
the infiltration
occurs along the white matter tracts.
Radiation is often
of limited benefit. Steroids help in palliation. Surgery, obviously, is
not possible, except for hydrocephalus,if any.
The prognosis is
dismal.
2) Astroblastomas:
They are rare tumors believed to arise
from astroblasts, the precursors of adult astroglia.
They are not well encapsulted, soft to
firm in consistency.
Histologically, there are prominent
elongated neoplastic cells with footplates forming pseudorosettes around
blood vessels. Limited mitosis may be present. Necrosis is seen in up to
70% of the tumors; but does not appear to have prognostic significance.
GFAP positivity may be present
occasionally.
Surgical resection is the primary
treatment. Role of radiotherapy and chemotherapy is not clear.
The prognosis is hard to predict; it
appears to be intermediate between that of astrocytoma and glioblastoma.
3)
Choroid gliomas:
They
are rare, have been reported in anterior third ventricle only.
Radiologically,
they mimic meningioma, and pit .adenomas.
They
are slow growing and present with hydrocephalus.
Histologically
they have glial features with ependymal differentiation, and there is
GPAF positivity. The cell of origin is not known.
The
treatment is surgery. The role of radiotherapy is not clear. The outcome
is generally favorable.
|
