|
These rare tumors have both glial and
neuronal elements.
They usually occur in children and the
young, and present with seizures.
They may mimic a low grade glioma,
radiologically.
Surgery is the primary treatment. The
overall prognosis is favorable.
WHO includes the following in this group:
|
Gangliocytoma,
Ganglioglioma (grade I or II),
& Anaplastic Ganglioglioma (grade III),
Dysplastic gangliocytoma of
cerebellum (Lhermitte-Duclos),
Desmoplastic infantile
astrocytoma/Ganglioglioma (grade I),
|
Dysembryoplastic
neuroepithelial tumor (DNT) (grade I),
Central Neurocytoma (grade
III),
Cerebellar Liponeurocytoma
(grade I or III), and
Paraganglioma of the filum
terminale (grade I)
|
Ganglioglioma:
This is an appropriate
designation, first introduced by Courville in 1930, as it
describes the most frequent form of this rare tumor group which
constitute less than 1% of primary brain tumors. They form 1% of all
intramedullary spinal cord tumors.
The mean age at diagnosis ranges from 16
to 25 years, with highest incidence in children, where they constitute 4%
to 10% of primary brain tumors. It has been reported in a 70years old.
Temporal and frontal lobes are the
favored sites. The spinal cord and the brain stem are next commonly
involved. Other less common sites are, optic nerve, chiasma, and tract.
An increased incidence in association with congenital anomalies, such as,
agenesis of corpus callosum and Down's syndrome is noted.
|
Although metastasis is rare,
leptomeningeal and subarchnoid spread have been reported.
Symptoms are related to the
location, with seizures and focal neurological deficits. Focal
neurological deficits with hydrocephalus occur in midline lesions. It
is one of the common causes of intractable epilepsy.
CT reveals an
iso to hypodense mass, with varying degree and calcifications and
cystic changes.
MRI reveals a
well circumscribed mass that is iso to hypodense on T1 and hyperdense
on T2 images. There is variable contrast enhancement.
|
|
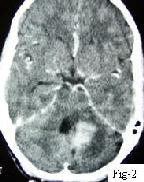
|
|
Ganglioglioma-contrast CT
|
|
|
Grossly, They are firm grayish,
avascular tumor with cystic components.
Histologically, glial and
neuronal elements are present in varying proportions. Glial elements
are mostly astrocytic.
In addition to anaplastic
gangliogliomas, other variants include, melanotic astrocytes,
neurocytoma, and pleomorphic xanthoastrocytoma.
Immunohistochemical staining
for synaptophysin, a neuronal cell membrane glycoprotein, helps in
diagnosis.
When a ganglion cell tumor contains
populations of small round cells with nuclear hyperchromasia, it is
labelled a ganglioneuroblastoma. These small cells proliferate
rapidly,
|
|
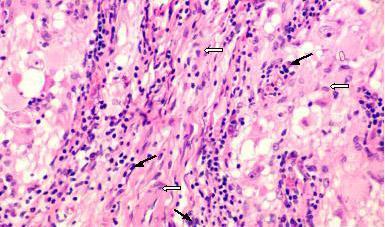
|
|
Ganglioglioma
(H&E): spindle
shaped glial (open arrows) and
ganglion
(black arrows) elements.
|
|
making
the prognosis worse. This tumor represents an intermediate step on a
spectrum from ganglioglioma to neuroblastoma.
Adenohypophyseal
neuronal choristoma arise from Ganglion cells within the anterior
pituitary gland (hence, it is not a brain tumor), most often within the
substance of a growth hormone producing pituitary adenoma. Such
neural choristomas (or pituicytomas) have been shown to elaborate growth
hormone releasing hormone (GRH).
Surgical resection is the mainstay of
treatment.
Post operative radiation is reserved for
lesions at eloquent areas, recurrence, and by some for anaplastic
variants. There is no substantial benefit with chemotherapy.
The prognosis is favorable with
10 year survival rate in excess of 90% of cases. Location of the tumor is
found to be a significant factor. Anaplasia, although debatable, does not
alter the prognosis significantly.
Recurrence rate is reported to be about
30%.The recurrence rate, reportedly, is 5 fold and 3.5 fold higher for
the brainstem and the spinal cord, respectively, compared with the risk
of recurrence for cerebral hemispheric tumors.
Desmoplastic Infantile Ganglioglioma
(DIGs):
These rare tumors, first described by Vandenberg
et al in 1987. Typically, they are supratentorial and detected in
infants with macrocrania and increased ICT and new onset seizures. They
may reach impressively large sizes and are characteristically found to
have contiguous leptomeningeal involvement, as reflected by the presence
of desmoplasia.
Imaging
of these neoplasms typically reveals a large, superficially located,
supratentorial mass with both solid and
cystic components. On CT, the solid component
of a DIG is seen as an avidly enhancing, iso to hyperdense region that is contiguous with the
meninges.
Cystic components underlie solid tumor
components. On MRI,
enhancement with
gadolinium is seen within the solid regions, which appear hypodense on T1 weighted
images, and of variable
density on T2 weighted images. Cystic areas appear hypodense and hyperdense on
T1 and T2weighted
images, respectively.
Grossly, it is a
firm large avascular mass with leptomenigeal extension.
Histologically,
both ganglionic and astrocytic neoplastic elements are present with dense
desmoplasia.
Surgery is the
primary form of treatment, with post operative radiation, as advocated by
some, in subtotal resections.
They generally
have favorable prognosis even in subtotal resection.
Gangliocytoma
or Ganglioneuroma:
Benign,
circumscribed, slowly expanding lesion that produces only local signs.
The majority of patients are under 30 and the temporal lobe is the
favored site. It may be solid or cystic and may show focal
calcification.
Microscopically,
it consists of mature but architecturally abnormal neurons, the glial
component being only sufficient as a stroma.
Hypothalamic
Neuronal Harmartoma: It is a specialized form of
gangliocytoma, first reported in 1934.
Clinically
symptomatic tumors may largely replace the hypothalamus and usually occur
together with endocrinopathy, e,g, obesity, diabetes insipidus or
precocious puberty. Most children present with precocious puberty before
the age of 3 years.
MRI
is the imaging of choice. T1 images show good resolution of the hamartoma
from the surrounding brain with no contrast enhancement. T2 may show an
iso or hyperdense lesion. They may be associated with midline deformities
including callosal agenesis, optic malformations, and hemispheric
dysgenesis.
Grossly,
the majority are pedunculated; some are sessile with a wider attachment
to the ventral hypothalamus.
Microscopically,
they consist of hypothalamic-type neurons, interspersed with glial
cells.
Treatment
consists of hormonal management similar to hormonally active pituitary tumors and surgery. A
pedunculated tumor is ideal for surgery, by a subtemporal approach.
Radiotherapy has not added to survival.
The
outcome is generally good, even with partial excision.
Dysplastic
gangliocytoma of the cerebellum (Lhermitte-Duclos): It was first reported in 1920 by Lhermitte & Duclos.
It is am extremely rare posterior fossa hamartomatous lesion, detected in
the 3rd and 4th decades of life, and occasionally in infancy. Although,
usually involving the cerebellar hemipheres, vermian involvement has been
reported.
Familial
affliction, and the presence of coexisting congenital anomalies, such as
megalencephaly, heterotopias, and polydactylia, has been described. An
association of has been made between dysplastic gangliocytoma and Cowden's
syndrome, an autosomal dominant disease, is characterized by the
presence of multiple hamartomas.
Clinically they
present with signs and symptoms of cerebellar dysfunction, or increased
intracranial pressure secondary to hydrocephalus.
CT reveals an
hypodense, nonenhancing enlargement of cerebellar folia and coexistent
hydrocephalus. There may be calcifications. MRI reveals the
characteristic striated pattern of dysplastic gangliocytoma. These
striations consist of alternating bands of hypo and isodense striations.
Grossly, it is a
mass composed of hypertrophied folia with myelination evident to the eye
in the outer part of the molecular layer.
Histologically, there is
disruption of cerebellar architecture with proliferation of ganglion
cells in the granule cell layer. There is thickening of the molecular layer,
widening of the granule cell layer, disappearance of Purkinje cell layer.
Neovascular proliferation may be present in areas with the highest
neuronal concentration.
Surgical
resection, and if necessary CSF diversion is the line of treatment. Some
recommend radiation in recurrence.
Dysembryoplastic
neuroepithelial tumor - DNT/DNET:
This rare tumor was described by Daumas-Duport
et al in 1988.
The histogenesis uncertain. It is
hypothesized to arise from external granular layer of the cortex.
The common Locations are cortical, and
temporal lobe, followed by frontal.
They are most common in children
and young adults, but can occur at any age.
Clinical Presentation is usually with
partial complex seizures, especially beginning before age 20 years.
Histological diagnosis is based on
presence of a specific glioneuronal element, consisting of
oligodendrocytes in a mucinous matrix in which neurons appear to float or
glial nodules associated with cortical dysplasia. Neuronal markers
(synaptophysin, neuronal
specific enolase) and glial markers
(GFAP, S-100) positive.
|
CT and MRI reveals a nodular
cortical lesion without edema. They may have megagyric or multicystic
appearance. Occasionally may enhance or contain calcifications. CT may
show calvarial remodeling.
Along with low grade gliomas,
and gangliogliomas they are grouped as a surgically curable cause of
intractable seizures.
Despite benign behavior, may
have high MIB-1 labeling index.
They are usually stable, even
over long periods, even after subtotal excision. Radiotherapy does not
appear to be of benefit. no data is available on chemotherapy.
Central Neurocytoma:
|
|
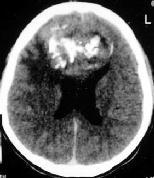
|
|
DNET-CT
|
|
These are rare intraventricular tumors
that occur in young adults with a male preponderance. They, probably,
represent a relatively well differentiated form of neuronal tumor. They
are frequently attached to the septum pellucidum or the walls of the
lateral ventricles. Extension through the foramen of monro into the third
ventricle is commonly noted. Extraventricular locations have been
reported.
The patients present with features of hydrocephalus,
and seizures in extraventricular ones. Spontaneous hemorrhage may be the
presenting feature in some.
CT and MRI typically reveal a variably
enhancing iso to hyperdense intraventricular mass with calcification and
associated hydrocephalus. Differential diagnosis include
oligodendroglioma, ependymoma, choroid plexus papilloma, subependymoma,
and intraventricular meningioma.
Histologically, they appear as sheets of
uniform cells in a streaming or 'Indian file' arrangement.
Electron microscopy reveals
neurosecretory dense core granules, neuritic processes, and areas
resembling synaptic junctions.
Immuno histochemistry is often necessary
for diagnosis. Synatophysin and neuron specific enolase reactivity is
detected.
Complete excision is the main stay of
the treatment. Role of radiation is debated.
Although thought to be benign, the
natural history is not clear.
Paraganglioma
of filum terminale:
It
is a disease of adults, unlike other neuronal tumors, and unlike other
paragangliomas (Paraganglia, elsewhere, have been shown to have
chemoreceptor roles with modulation of respiratory and cardiovascular
function), they are non functional. There is male preponderance.
They are rare. Almost always, the location is at L2/3, just distal to conus.
They are slow growing and expand in all directions involving multiple
roots at cauda equine.
Clinical
presentation is like any other degenerative multiple disc degenerative
diseases. They may present with cauda equine syndrome. Diagnosis is
usually made after MRI. It is intradural hyperdense lesion with
dural attachment.
Surgery
is the treatment, with careful protection of rootlets. The tumor is very
vascular. Invariably, the tumor is suspended by a feeding artery and its
accompanying vein at the cephalic pole of the tumor. Once this artery is
secured, the tumor becomes avascular. Piecemeal excision is not advised.
Few rootlets need to be sacrificed.
Histologically,
there are uniform polygonal cells with pale eosinophilic granular
cytoplasm. All reported intradural cases are benign.
Prognosis
is good, with no recurrence after total excision. Role of radiotherapy,
in subtotal excisions, is not clear.
|
