|
Craniosynostosis is
the premature partial or complete ossification of one or more of the
sutures separating the membranous bones of the skull. The incidence
of craniosynostosis has been estimated to be 1
in 2,500 live births.
Primary,
or simple, craniosynostosis is ‘single- or
multiple/compound’ suture synostosis in children who are otherwise
neurologically normal, and almost always
present prenatally except in some with cranio
facial syndromes of Crouzan's, Apert's and Carpenter's type in whom progressive
postnatal closure may occur. Synostosis that occurs as part of a
syndrome of complex congenital malformations is frequently called
complex, or syndromic, craniosynostosis.
Secondary stenosis may occur in children with metabolic
disorders, and hematological disorders.
Rickets, Hyperthyroidism, Thalassaemia, or Mucopolysaccharidosis may be associated. It can also
occur as the consequence of lack of growth at the suture lines because of
microcephaly, encephalocele, or shunted hydrocephalus.
PATHOGENESIS:
The cranial vault develops intramembranous bone formation
between the periosteum and the dura. This process begins during the 6th
week of embryonic development. The posterior fontanel closes at 3 mths of age and the anterior one, at 8mths of age.
The bones ossify by the end of first year. The skull growth ceases by
10-12 yrs of age. The ossification of the
cranial sutures occurs by the 4th-5th decade. The base develops from a
series of primordial cartilages that undergo ossification. The growth of
the base proceeds at a slower rate.
The skull contains two types of sutures- syndesmoses and synchondroses.
Syndesmoses occur in the vault and consist of fibrous tissue
interposed between bone surfaces. Synchondroses
occur in the skull base from a bar of cartilage. The role of sutures is
unclear.
The normal infant's skull is oval shaped and widest
posteriorly. The growth occurs in the direction perpendicular to the
suture lines, the direction of least resistance.
In craniosynostosis the deformity
is related to the suture involved and the effects of increasing growth of
the brain upon the unfused skull plates. The craniosynostoses
comprise a heterogeneous group of disorders of multifactorial origin.
Most
cases of simple craniosynostosis, are sporadic. The
cause is unknown. Many theories exist.
Genetic determinants play a part in some coronal and pansynostosis, particularly where there is an
associated craniofacial abnormality. Some cases are inherited. Affected
families may have members with involvement of different sutures, and, in
the case of familial coronal synostosis, family members may have
unilateral or bilateral involvement. Mendelian disorders may cause simple
or complex craniosynostosis, but craniosynostosis associated with chromosomal
aberrations is usually part of a syndrome.2 Approximately 27
different chromosomal aberrations have been reported for craniosynostosis, and more continue to be described
each year. Dominant inheritance is more common than recessive
inheritance. Most cases of simple craniosynostosis,
however, are sporadic.
Abnormal tensile forces along the dural
tracts running from the cranial base to the vault induce premature suture
fusion. These dural tracts consist of tentorium
cerebelli, the falx and dural
bands from the lesser wing to the vault.
Some blame the fetal head constraint in utero. The
periosteum overlying the suture may be involved.
Scaphocephaly: (Boat
Skull)
|
Premature
fusion of the sagittal suture results in boat skull. The skull grows parallel to the fused sagittal
suture, the skull becomes elongated as the frontal and occipital bones
compensate for the restricted lateral growth of the parietal bones
resulting in frontal bossing. The sagittal suture may become prominent
as a ridge. 2% of the cases are familial, and about 80% are males.73
to 80 per cent of scaphocephaly patients are
males.
Plagiocephaly:
(Asymmetric Skull)
|
|

|

|
|
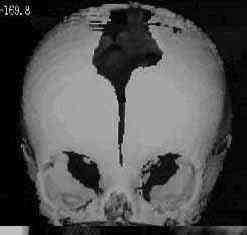
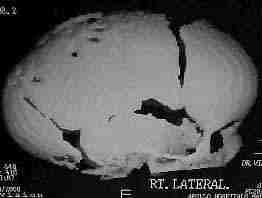
Scapocephaly-AP
|
Scapocephaly-Lat
|
|
|
There is premature closure of one coronal suture resulting
in under development of the forehead, supraorbital ridge and anterior
fossa on the ipsilateral side. Radiologicaly,
in addition to the obliteration of the coronal suture, there is a
deformity of the orbit resulting from elevation of the ipsilateral
sphenoid ridge, commonly described ‘harlequin orbit'. This type of
synostosis is more commonly associated with a clinical syndrome than is
scaphocephaly, but the majority of cases
remain sporadic. Males are affected twice as often as females.
If
it is inherited in families, other family members tend to have
bilateral coronal synostosis, suggesting that the two conditions may
have a similar genotype. The lambdoid
synostosis produces a posterior plagicephalic
skull as a result of the flattening of the parietooccipital
region ipsilateral to the fused suture, and may be seen as a
part of a complex craniofacial syndrome.
|
|

|

|
|
Plagiocephaly-AP
|
Plagiocephaly-Lat
|
|
|
Brachycephaly:
(Short Skull)
It is a broad short skull due to bicoronal
synostosis and brain growth ipsilateral to the coronal sutures. Radiologicaly, there is bilateral harlequin’
orbits. It is the commonest.
|
|

|

|
|
Brachycephaly-AP
|
Brachycephaly-Lat
|
|
|
Trigonocephaly:
(Triangular Skull)
There is premature fusion of the metopic suture. The
transverse growth of the forehead is compromised. It is associated with
orbital hypotelorism. The resultant shape
gives rise to triangular shaped skull. It may be associated with
Christian syndrome II, an X-linked, semidominant
syndrome consisting of hypertelorism, clinodactyly, vertebral anomalies, and imperforate
anus. Metopic synostosis is one of many different suture synostoses
seen in Carpenter's syndrome.
|
|

|

|
|
Trigonocephaly-AP
|
Trigonocephaly-Lat
|
|
|
Oxycephaly: (Towering skull)
Simultaneous
fusion of multiple sutures may produce a conical-shaped head and is
seen in 5 to 10 per cent of primary craniosynostoses.
It is the result of pansynostosis
where all the sutures are prematurely fused and the skull assumes a
towering shape-towering skull. Increased ICT is more often present.
The deformity is progressive over the period of maximal
brain growth, during the first 12mths,less
progressive during the 2nd year and reaches its maximum at the age of
2yrs and unchanged after that time. This condition is heterogeneous in
origin and may be seen in Crouzon's syndrome.
|
|

|
|
|
Oxycephaly-AP
|
Oxycephaly-Lat
|
|
ASSOCIATED DISORDERS:
The sagittal synostosis is rarely associated with other
abnormalities.
About 30% of the unilateral coronal synostosis and 60% of
those with bicoronal synostosis have other
congenital abnormalities, such as syndactyly and cardiac anomalies.
The patients with craniofacial syndromes of the Crouzon’s, Apert’s or Carpenter’s variety almost invariably have
associated craniosynostosis.
|
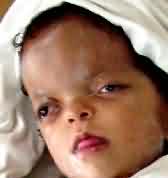
Apert's syndrome: There is brachycephaly with
midface hypoplasia. There is a unique hand malformation.
Both hands are affected equally, as are the feet. This unusual
variation of syndactyly can be used to identify Apert's
from other similar syndromes.
Usually, cases are sporadic, but the syndrome may be inherited in an
autosomal dominant fashion.
|
|
|

|

|
|
Apert's
|
Apert's
feet syndactyly
|
Apert's
hands syndactyly
|
|
Crouzon’s syndrome: It is commoner. There is brachycephaly with midface hypoplasia and shallow
orbits with proptosis. The skull base is implicated, and sphenofrontal synostosis is a factor involved in
producing exophthalmos. Maxillary hypoplasia, the characteristic facial
deformity, is a more important cause of exophthalmos. In 5 per cent of
cases, the calvaria is normal. Hydrocephalus
is more common in Crouzon's syndrome than in simple craniosynostoses.
Unlike in Apert’s there is no
syndactyly.
This
condition is inherited in an autosomal dominant fashion, but there is an
equal incidence of sporadic cases, which probably represent new
mutations. Penetrance is high, although severity is variable. Within the
same family, members tend to have similar facial deformities but variable
calvarial deformities. This phenotypic
heterogeneity makes genetic counseling difficult.
Variations of this type of craniofacial dysostosis
are seen.
In
Carpenter's syndrome, the calvarial deformity
is variable and is less striking than the facial and digital anomalies.
Mental retardation, cardiac anomalies, and hypogonadism are also common.
Saethre-Chotzen syndrome is a
relatively benign condition. The features of this condition include brachycephaly or plagiocephaly,
facial asymmetry with hypertelorism and orbital
dystopia, low frontal hairline, ptosis, and mild maxillary hypoplasia.
Syndactyly is usually mild, and mental retardation is much less common.
Pfeiffer's
syndrome is characterized by tall, broad skull, hypertelorism,
slanted palpebral fissures, and broad thumbs and toes; mentation is
usually normal.
CLINICAL FEATURES:
1) The skull deformity is obvious, so also are the
associated facial deformities.
2) Mental retardation can occur due to associated brain
malformations, or increased ICT. It is unknown in single suture
synostosis.
3) Hydrocephalus is infrequent. The mechanism is not known.
4) Increased ICT is rare in single synostosis. It is more
frequent in the first 2 yrs.The pressure tends
to normalize at six years of age. The pressure may take months to come
down after decompressive surgery.
DIAGNOSIS:
The plain x-rays and CT scan in axial and coronal
planes, with bone windows, will give adequate information and also reveal
brain malformations and the presence of increased ICT, if any.
|
|
|

|

|
|
|
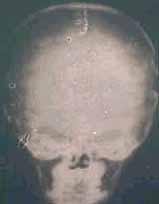
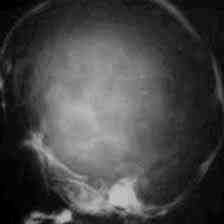
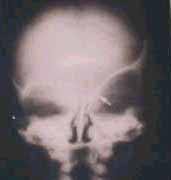
X- ray(AP)-brachycephaly
|
X- ray(lat)-brachycephaly
|
X-Ray(AP) scapocephaly
|
X-Ray(lat) scapocephaly
|
X-ray-Harlequin orbit-Plagiocephaly
|
|
3D CT adds to the clarity.
MRI shows any associated cerebral
malformations better.
Positional deformity, due to birth molding, may be
diagnosed by the presence of patent sutures and absence of any
orbital deformity.
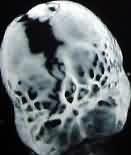
In microcephaly, the brain fails to grow and the
head remains small. The fontanels close early. The x-rays reveal a
thick vault, and the frontal vault is disproportionally small. X-rays
do not show any silver beaten appearance or sellar
changes.
|
|
|
|
|
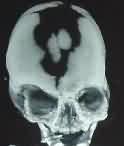
Apert's -3D CT
|
3D CT- craniolacunae
|
|
MANAGEMENT:
The management is by a multidisciplinary team comprising of
the neurosurgeon, the plastic surgeon, the pediatrician, the
ophthalmologist, dental surgeon, anesthetist and the social worker.
The aim of surgery is
1) to allow normal brain growth.
2) to correct increased ICT.
3) to achieve cosmetically acceptable head.
4) to protect the eyes.
The ideal time for surgery in the absence of increased ICT,
is 3 - 6mths of age to give the growing brain the ideal surroundings and
also to facilitate normal shape to the skull. Surgery for cosmetic
reasons, especially in coronal synostosis, can be performed at a later
age.
A generous craniectomy with
extension to adjacent sutures is the basic procedure. Frontal and orbital
advancement techniques are needed in coronal synostosis. Dural-pericranial stitches or use of tantalium
foils at the site of craniectomy is advised to
prevent reossification.
The associated facial deformities can wait until about 5 yrs of age to allow the roots of the teeth to descend
and permit safe correction of the maxilla.
|
|

|
|
|
|
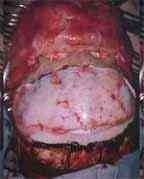
Frontal bones exposed
|
Excised supraorbital ridge
|
supra orbital ridge advanced
|
frontal bones repositioned
|
|
Scapocephaly-3D-CT-post-op (AP)
|
Scapocephaly-3D-CT-post- op ( lat)
|
In metopic synostosis, the surgery is for cosmetic reasons
and involves remodeling of the frontal bone and supralateral
orbital advancement. The triangular forehead is removed and recontoured to the appropriate shape or bone may be
taken from another area of the skull to replace this bone. The entire
bony supraorbital bar is removed and reshaped with supralateral
orbital advancement to restore the normal brow contour. Wires or
micro-plates are used to secure the frontal bar to the facial bones and
maintain the normal contour.
In bicoronal synostosis, both
frontal bones are reconstructed. The supraorbital bar is completely
removed, reshaped, and straightened. The supraorbital bar is advanced and
lowered as necessary on the affected side and rigidly fixed in position
to the face. The reconstructed forehead is then secured to the
supraorbital bar. In unicoronal synostosis, the
surgery is for cosmetic reasons and restricted to the involved side.
|

|
|

|

|
|
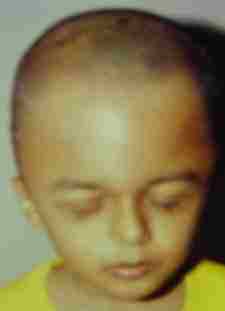
Crouzon's-pre-op
|
Crouzon's-post-op
|
Crouzon's-10yrs later-AP
|
Crouzon's-10yrs later-Lat
|
|
In lambdoid
synostosis, the surgery is purely for cosmetic reasons and consists of
extensive posterior skull craniectomies with
remodeling of both sides of the occiput and posterior advancement of
the affected side.
In oxycephaly, the aim is to relieve the increased ICT and
the surgery involves multiple craniectomies
of the fused sutures.
Restenosis is well
known and may require resurgery.
|
|