|
Craniopharyngiomas are the
commonest of a heteregenous group of tumors that have in common their
congenital origin and slow rate of growth. They have always been one of
the most controversial problems of neurosurgery concerning their origin,
optimal therapy, operative approach, and response to radiation.
Craniopharyngiomas are still one of the most challenging tumors for
neurosurgeons. Despite the significant technical advances in
microneurosurgery, radiation therapy and endocrinology, the controversies
have not abated.
Controversies apart, for all
practical purposes, they are better considered as true neoplasms, rather
than an hamartoma or a cyst. Rightly, WHO has listed
them as sellar tumors, although purists may not agree.
Incidence:
Craniopharyngiomas
constitute between 2.5-4% of all intracranial tumors. There are 0.5-2 new
cases per million populations occurring each year. Almost 50% are in
adults. Craniopharyngioma is the most common tumor of non-glial origin in
children representing 54% of all suprasellar tumours in childhood and 20%
of those in adults.
They may present at any age
with preponderance in childhood. The highest peak of incidence is between
the ages of 5 and 10, a moderate peak exists among adults in the 4th and
5th decade. The tumor occurs with equal frequency in both sexes in all
ages.
Pathology:
Since the anatomical
structures in the suprasellar region are normally devoid of epithelial
cells such as those seen in craniopharyngiomas, the nature and source of
these cells have long been the subject of intensive investigation. In
1899, Mott and Barrett stated that Craniopharyngiomas may
arise from the hypophyseal-pharyngeal duct or Rathke’s pouch. In
addition, Erdheim demonstrated in 1904 that these are epithelial
tumors arising from embryonic squamous cell rests of the incompletely
involuted hypophyseal-pharyngeal duct located in the pars tuberalis and
along the pituitary stalk. This theory continues to be widely accepted.
However, since squamous cell rests are rarely found in neonates, and
their presence increases with age, some argue that these tumors are not
originated from those embryonic rests, but may arise by metaplasia of
normally developed anterior pituitary cells.
Craniopharyngiomas are
histologically benign tumors of epithelial origin involving primarily the
sellar region with a tendency to adhere to vital neural and vascular
structures. Although usually well circumscribed and extracerebral in
location, they often extend into the neighboring brain tissue evoking a
variable degree of glial reaction. They may also be strongly adherent to
major arteries and cranial nerves at the removal difficult and sometimes
impossible. Postoperative complications and tumor recurrence are
frequent. Consequently, this benign tumor is often malignant in clinical
behavior.
Craniopharyngiomas are mostly
located in the suprasellar region and grow by expansion into the
hypothalamus and third ventricle upwards, growing into the sella, down
between the clivus and brainstem, or even into the cerebelloprontine
cistern. There are some rare primary locations, such as: intrasellar,
within the third ventricle, nasopharyngeal, craniobasal, and pineal.
Occasionally they may be enormous. Occlusion of the CSF pathways with
subsequent hydrocephalus occurs in 15 to 30% of the cases.
The macrosocpic appearance
of craniopharyngiomas is wholly cystic in 34%, purely solid in 23% and
mixed in 43%, thus about 77% tumors are cystic. The fluid in these cysts
is oily yellow, brown, or greenish. This solution contains variable
amounts of protein with suspended cholesterol crystals which are
birefringent to polarized light is viscosity varies from watery to
viscous. Cyst walls may be transparent membranes or dense, tough
structures. Calcification is found in about half of adult
craniopharyngiomas and in almost all children.
Histology:
Craniopharyngiomas are generally composed of mature epithelial cells with
an external layer of high columnar epithelium, a variable portion of
polygonal cells and a central network of epithelial cells, without any
sign of malignant heterogeneity, cellular atypia or mitoses. This
structure is supported by variable amounts of loose growth of these
tumors is not neoplastic, but results from simple cellular proliferation
of the epithelium, desquamation, degeneration and colliquation of the
epithelial cells with active sequestration of cystic fluid. Regressive
changes in the epithelial cells include cellular liquefaction resulting
in cyst formation or deposition of a keratin-like material or calcium
salts resulting in calcification.
There are two basic craniopharyngioma
variants, the adamantinous and squamous papillary type. There is a
correlation between the histopathological, clinical, and prognostic
features of the two histological types. The behavior of childhood
craniopharyngioma is distinctly different from that in adults.
|
Adamantinous
craniopharyngiomas (48% of all cases) develop mainly in childhood, but
may occur in all ages. These are cystic, calcified tumors which are
prone to recur even after radical surgical removal and have a worse
overall outcome. They are characterized by a layer of palisading
columnar cells resting on a thin basement membrane with loose
aggregates of stellate epithelial cells in the middle. Central
degenerative changes are frequent, leading to cyst formation, keratin
nodule development and calcification.
Squamous papillary
craniopharyngiomas (33%) occur predominantly in adults. They are
chiefly solid, non calcified lesions with little tendency to recur
after total removal and thus a significantly better overall outcome. Microscopically,
squamous epithelial cells form solid nests or sheets embedded in a
loose connective tissue stroma.
Both the squamous
and adamantinous tumors may contain solid and cystic areas,
|
|
|
though their
proportion is usually different in the two types.
Beside these two
forms, a third, mixed type (19%) has been described. Because of
the frequent association of admantinous tumors with large squamous
epithelium-lined cysts, many investigators have suggested that
craniopharynigomas represent a spectrum of a single group of tumors,
with a range of characteristics from the purely adamantinous type
through a mixed variety to the squamous papillary type.
|
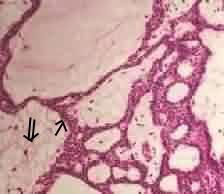
Adamantinous
craniopharyngioma (H&E): anastomosing cords of
predominately basloid epithelial cells(arrow) enclosing
areas of loose collagenous matrix (double arrow)
|
|
The histological
differences between adult and child hood craniopharyngiomas might
indicate separate origins. It has been argued that the pediatric
(adamantinous) group represents an embryogenic disorder and the adult
(squamous) tumor arises from metaplastic cells.
Craniopharyngiomas
do not invade neural tissue, but often cause an extensive glial
reaction at the interface, which is particularly dense around small
finger-like tumor projects toward the hypothalamus. Traction on this
tenacious attachment may lead to hypothalamic infarction and thus
preclude safe total removal of the tumor. In other cases this glial
envelope may provide a plane in which to dissect between the tumor and
the hypothalamic structures making total surgical removal possible.
Both features can be present in the same tumor.
Similarly,
craniopharyngiomas are frequently strongly adherent to major arteries
and
|
|
|
cranial nerves at
the base of the brain. The most tenacious attachments are to the stalk,
and the arteries of the anterior portion of the circle of Willis.
Adherence to these structures can vary from mere approximation to
persistent attachment preventing total removal in some cases.
|
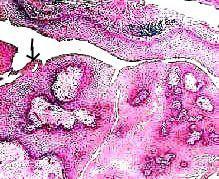
Papillary
craniopharyngioma( H&E): squamous
epithelial cells(arrow)
form solid nests or sheets embedded in a loose connective tissue
stroma.
|
Symptoms and signs:
The clinical signs are due
to the location of the tumor, though adults and children may have
different clinical syndromes. The multiplicity of possible directions of
growth of these tumors is mirrored by a corresponding variation in the
clinical pictures they may present.
Children:
Since these tumors may reach a large size before causing symptoms in
children, they usually present with signs of increased intracranial
pressure mainly due to occlusion CSF pathways and secondary hydrocephalus
or due to the size of the cyst. Disturbances of hypothalamic-pituitary
function such as growth failure, pituitary dwarfism, hypogonadism,
delayed puberty, diabetes insipidus, genital dystrophy are also
characteristic in about 93%. Visual loss and visual field defects are
usually found at examination, though children tolerate remarkable degrees
of visual loss without complaint.
Adults:
Visual impairment and visual field defects (mostly bitemporal hemianopia)
are characteristic complaints in adults, but any combination of visual
symptoms can be present, similar to those of a pituitary adenomas.
Papillioedema is uncommon, but optic atrophy is frequent in both age
groups. Progressive visual failure requires urgent treatment in many
cases. Psychiatric symptoms include personality changes, dementia memory
loss, drowsiness, confusion, depression, hyperphagia, aggression which
can develop in association with obstructive hydrocephalus and with
hypothalamic dysfunction. Patients frequently (85%) present with
endocrinopathies such as menstrual irregularities, loss of libido,
diabetes insipidus, hypothyrodism, hypodrenalism, and obesity. Symptoms
suggesting intracranial hypertension often prompt patients to seek
medical attention. Spontaneous rupture of craniopharyngioma cysts is
extremely rare; only a few cases have been reported causing chemical meningitis
with temporary alleviation of headache and improvement in visual
disturbance.
Differential
diagnosis:
In Children: Hypothalamic
and optic gliomas, Germinomas, Hamartomas, Teratomas, Pituitary adenomas,
Histocytosis X, Chordomas, Ectopic pinealomas, Primitive neuroectodermal
tumours
In adults: Pituitary
adenomas, Rathke’s cleft cyst, Epithelial cyst, Meningioma, Epidermoid,
Dermoid, Arachnoid cyst, Cavernous angioma, Colloid cyst, Tuberculoma,
Metastasis, Internal carotid artery aneurysm.
Diagnosis:
There are a number of
neuroimaging methods to reveal the craniopharyngiomas surgically
important features as well as to differentiate them from other possible
suprasellar masses.
Plain skull X-rays show
pathological changes in most of the adult and almost all of the pediatric
cases. Tumor calcification is found on X-ray in 85% of the pediatric and
405 of the adult cases. Other signs include enlarged sella, bony erosion
of the sella, dorsum, or clinoids, and signs of increased ICP.
CT scan shows
the extent of the lesion, distinguishes between solid, cystic and
calcified components and demonstrates hydrocephalus. The appearance of
the cyst fluid is variable. It is usually of low density, but can be
isodense or hyperdense if it contains sufficient protein and suspended
calcium salts. The solid portions of craniopharyngiomas are either
isodense or slightly hyperdense. Contrast administration mostly causes
intensive heterogeneous enhancement. Ring-shaped contrast enhancement can
sometimes be seen around cysts. Coronal scanning helps to identify
intrasellar extension, relation to the third ventricle and impingement of
tumor on the basal brain parenchyma.
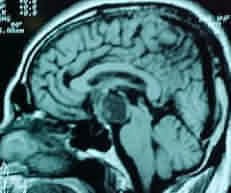
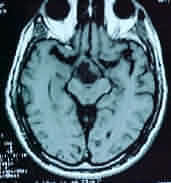
MRI is
superior to CT in demonstrating the general configuration of the tumor
mass, with us relationship to the ventricular system, major nerves and
cranial arteries. It characteristically shows a heterogeneous suprasellar
mass containing cysts and empty holes correlating with calcification.
Cysts usually appear bright on T2-weighted sequences. The signal intensity
of the fluid varies on T1-weighted images from hyperintense to hypotense,
reflecting the heterogeneous contents of cysts. Cysts with a low protein
content are indistinguishable from cysts of other etiology. Calcification
produces a low signal on MR images, which are less specific in this
respect than CT scans. Solid tumor components present a less well-defined
margins compared to pituitary adenomas. These areas intensively enhance
with gadolinium contrast on T1 weighted studies.
|
|
|
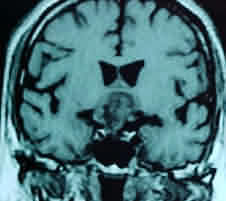
|
|
Craniopharyngioma-MRI- sagittal
|
Craniopharyngioma-MRI- axial
|
Craniopharyngioma-MRI- coronal
|
Angiography has
become redundant these days.
Management:
The treatment of cranipharyngioma
remains controversial, reflecting the difficulties in management as well
as the heterogeneity of these tumors. 15 to 30% of the patients require
attention to associated hydrocephalus.
In most cases,
cranipharyngiomas can be removed totally or subtotally even when they
have reached a great size. There is little doubt that an initial total
tumor excision yields the best long term outcome if it can be achieved
without neurological sequelae. Some workers regard surgery of
carniopharyngiomas as merely palliative and believe that subtotal or
partial resection followed by radiation therapy should be the rule.
While total removal may be
associated with significant endocrine alterations and hypothalamic
disturbances, subtotal removal results in higher rates of recurrence.
Most reviews conclude that
total excision should be attempted whenever feasible, and subtotal
removal with adjuvant radiation should be done when total resection is
dangerous. In cases of recurrence, radical tumor removal, subtotal
resection and/or irradiation are again the usual treatment modalities.
Adjunctive therapy should be
reserved for recurrent or predominantly cystic cases.
Strict
endocrinological evaluation and hormonal replacement regiments are
essential in all patients in both the immediate and long term
postoperative period. (refer
to pituitary adenoma)
Multimodality therapy is
often necessary over the course of a patient’s lifetime. Due to the
diverse nature of craniopharyngiomas, therapy must be individualized to
the particular clinical problem by following the natural history of each
case.
Surgical approaches:
There are several operative
approaches to craniopharyngiomas. Each has its own advantages and
disadvantages. The side of the craniotomy is determined by considering
various factors such as visual acuity and field defect, lateralization of
cyst, calcification and/or solid mass, lateralization of the compromised
hypothalamus, and the preference of the surgeon.
Unilateral
operations are preferable.
a) The pterional
(frontotemporal) approach provides access to virtually all parts of
even very large tumors. It is the shortest route to the parasellar
region and allows good visualization of the retrosellar area; but
visualization of the contralateral optic nerve is limited.
b) The
unilateral or bifrontal subfrontal approach allows good visualization of
optic nerves, chiasm and ipsilateral carotid artery, making the presellar
anatomy easily understandable. On the other hand, it does not give
good access to the area beneath the ipsilateral optic nerve and tract,
and the region of the third ventricle.
c) The transcallosal
approach uses a fronto-parasagittal craniotomy. This route is
preferable when the tumor is within or in the region of the third
ventricle and the tumor pushes the anterior hypothalamus forwards.
In these situations a basal approach would result in damage to the
hypothalamus before the tumor has been reached. This approach offers good
visualization of both walls of the third ventricle. Anterior corpus
callosum needs to be divided and there is a risk of bilateral fornix
damage.
d) The transcortico-ventricular
approach is associated with a cortical incision, requires the
presence of hydrocephalus, and allows limited visualization within the
third ventricle. This offers good visualization of the ipsilateral
foramen of Monro.This technique has been largely abandoned.
e)
Transsphenoidal surgery avoids craniotomy and is best reserved for
patients with enlarged sella and intrasellar infradiaphragmatic,
primarily cystic craniopharyngiomas. Enlargement of the sella is of
primary importance when using this approach, since it implies that the
tumor took origin under the diaphragma sellae, thus it is not attached to
the suprasellar structures such as the chiasm or hypothalamus, making
total removal possible. This approach is also used to drain cystic
tumors, and as part of staged surgery in partially intrasellar tumors.
Tumor dissection
uses combinations of routes.
The interoptical
route, between the optic nerves, gives an excellent view of both carotid
arteries. The optic carotid pathway lies between the carotid artery
and the optic nerve and tract.
Intraventricular
craniopharyngiomas can be approached through the lamina terminalis.
This pathway permits visualization of the anterior third ventricle and
gives access to supra- and retrochiasmatic masses, but carries the
highest risk of hypothalamic damage..
The lateral and
clival tumor surface can be approached between the carotid
artery and oculomotor nerve.
The intracranial
transsphenoidal pathway requires drilling partially away the
tuberculum sellae and the anterior wall of the sellae and opening the
sphenoid sinus without disrupting its mucosal membrane. This can be
useful when the tumor has a deep intrasellar extension, and when the
chiasm is prefixed.
In cases of
hydrocephalus, primary tumor removal usually re-establishes normal CSF
pathways avoiding shunting. However, when the hydrocephalus is
pronounced ventricular shunting may be performed as a first step.
The surgeon
should be familiar with these approaches and should use a combination of
these techniques as necessary.
Surgical technique:
Once all the major neural
and vascular structures are identified, each of the possible routes of
approach is evaluated and the greatest possible tumor surface is exposed.
Since these are subarachnoid tumors, the preservation of the plane
between the tumor capsule and the arachnoid is essential for safe and
total tumor removal. Besides general brain protection, the entire
intradural area is lined with cotonoids to prevent the spread of the
irritating crystalline content of cyst or solid tumor on to the brain or
into the CSF, as it may lead to an aseptic meningitis or hydrocephalus
postoperatively.
The optic nerves and chiasm
are most often found to be stretched anterosuperiorly by the pressure of
the tumor. After additional manipulation, any residual function may be
lost. Upwards displacement of the optic nerves against the sharp upper
edge of the optic foramen invariably results in visual loss. Before
causing more damage to the optic nerve, the foramen should be
decompressed at an early stage by partly removing its bony roof and
opening its dural sleeve.
The usually severely
compressed pituitary stalk and infundibulum may be found displaced
behind, above or lateral to the tumor. Preservation of the stalk, in
cases when it is not destroyed by the tumor, may occasionally be
possible. Although it may be functionally disconnected by the tumor
compression and surgical manipulation, a remnant of stalk reaching from
the medial eminence to the pituitary gland may serve as a matrix upon
which the pituitary portal system may reform.
Aspiration of any cyst is
the first step. The arachnoid covering of the tumor is carefully opened.
Entering the tumor with a microsuction tip will further decompress the
tumor. As the tumor mass decreases, more of the arachnoid capsule may be
exposed by dissecting in the plane around the tumor. Craniopharyngiomas
tend to get their arterial blood supply from the vessels of the anterior
circle of Willis. Direct branches from the carotid, the anterior cerebral
and the posterior communicating arteries have been described. Within the
sella turcica the tumor may be supplied by small perforating branches
directly from the cavernous portion of the internal carotid artery. They
do not receive blood from the posterior cerebral or basilar arteries
unless the third ventricle is invaded. These feeders may be coagulated
and divided. The interior of the tumor is entered and the solid portion
removed piecemeal. As the tumor is progressively gutted, the capsule may
be gradually resected.
Every effort should be made
to preserve the arachnoid capsule so that leaving behind torn-off bits of
the capsule is avoided. Even the tiniest scrap left behind can cause
tumor recurrence. Using small angled dental mirrors is recommended to
gain vision to the undersurface of the optic apparatus and hypothalamus.
Use of endoscope is a better option.
Tenacious adhesions of tumor
to main and perforating arteries, usually on the anterior circle of
Willis, are the most common obstacle to achieving total removal. This
mesenchymal reaction to the craniopharyngioma capsule appears to be
denser than the glial scar that forms underneath the chiasm or
hypothalamus.
Intraoperative arterial
damage should be avoided during dissection of these adherent parts.
Large solid calcified
portions can be difficult to break up and remove as they may pose a risk
to the neural and vascular structures as the jagged pieces of calcified
material are delivered past them. Calcified tumors, particularly those
adherent to or enclosing vital structures, have a lesser chance of total
removal.
In tumors that infiltrate
the hypothalamus it may be found that in some cases these structures can
be easily preserved, while in others it is impossible to distinguish
between the normal hypothalamus, the gliotic tissue and the tumor. The
ease of removal and risk of dissection can vary accordingly. A dense,
invasive finger-like tumorous and gliotic tissue with diffuse adhesions
of the hypothalamus renders it impossible to follow a plane of cleavage.
This situation limits the total respectability of these tumors.
Operative
complications:
a) Preoperative endocrine
deficiencies are irreversible in most cases. Surgical intervention,
however, often leads to additional endocrine disorders. After
craniopharyngioma surgery most patients have temporary or permanent
disruption of neurohypophyseal axis function even if function was normal
preoperatively. Among the various endocrine abnormalities, the loss of
ADH and ACTH will be the most important in the postoperative period.
Diabetes insipidus monitoring and therapy, and prophylactic treatment
with corticosteroid are essential. Long term endocrine follow-up with
appropriate replacement therapy is required with special attention in
children.
b) Surgical manipulation of
these tumors may result in hypothalamic dysfunction. Damage to the
hypothalamic nuclei and varying range of deficits such as sudden death,
alterations of consciousness ranging from somnolence to coma, water and
electrolyte imbalance, loss of thirst, hyper-, hypo-,or poikilothermia,
cardiac disturbances, hyperphagia, obesity, insomnia, hypogonadism,
growth failure.
c) Disturbance of
hypothalamic connections to the thalamus, limbic system, frontal lobes
and other cortical areas may explain some of the psychiatric and social
problems seen following treatment of cranipharyngiomas. Injury in this
area, from surgical or less frequently from irradiation treatment, will
result in problems common among these patients: confusion, short term
memory loss, mutism, emotional and sexual immaturity, psychic imbalance,
hyperphagia and aggressiveness.
d) Visual impairment is a
relatively common surgical complication but can be minimized with
technique. Direct surgical manipulation of the stretched optic nerve,
chiasm and tract or their vascular supply may result in additional visual
loss. Its reported incidence ranges from 1 to 17%. The degree of
preoperative visual loss and its duration (optic atrophy) are the most
important factors in determining postoperative visual status.
e) Three types of vascular
lesions have been described after craniopharyngioma removal. The first is
fusiform dilatation of the internal carotid artery which may develop due
to the weakness of the arterial wall following dissection of the adherent
tumor from the adventitia, or due to the injury of the vasa vasorum.
Secondly ischemic symptoms may develop at sites distant from the
operative field which is thought to result from the development of
vasospasm related to the operative manipulation to the vessels, to the
blood appearing in the subarachnoid space or possibly to some chemical
substances of the spilled cyst fluid. Finally, in cases of massive
hydrocephalus, intraoperative ventriculostomy can cause sudden collapse
of the cortex with subsequent stretching of the cortical and bridging
veins and multiple venous infarcts over the surface of the cortex.
f) CSF rhinorrhoea and
meningitis are the most common complications of the transsphernoidal
approach.
g) Other possible
postoperative complications such as cranial nerve palsies, epilepsy and
hemiparesis are related to the location and not the nature of this tumor.
Operative results:
At present, overall
mortality for total removal of craniopharyngiomas is under 10% in
experienced hands.
There is a strong
correlation between the size of the tumors and the outcome. Tumors
smaller than 4 cm are usually totally removed with good to excellent
results, while total removal of large (>4cm) tumor is associated with
significantly higher morbidity and mortality. These facts stress the
importance of early diagnosis and treatment of craniopharynigomas.
Virtually every series of
craniopharyngiomas has reported recurrences of tumor even after a
macroscopic total removal. Although these tumors are known as slow
growing tumors, they tend to recur early because of the capacity for
rapid growth of the cystic portions.
In different series, the
majority of recurrence was noted within 2 years of surgery, though
recurrences have been reported as late as 30 years after operation. In
one study, 52.5% recurred within one year, 75% within 3 years and 85%
within 5 years.
Reoperation and/or adjuvant
irradiation are recommended in cases of symptomatic recurrences. Surgery
of recurrent craniopharyngiomas has worse outcome in all series. The reported
mortality rates are much higher, being between 12 and 38% (Samii, 1995).
Recurrent tumors are more tightly adherent to vascular and neural
structures, making total excision more dangerous and therefore
impractical in many cases.
Yasargil
(144 cases, 1990) achieved total primary removal of craniopharyngiomas in
90%. Primary radical excision without additional radiotherapy was
associated with a good outcome in 76.8%, morbidity of 13.4% and an
overall mortality rate of 9.8%. The recurrence rate was zero in the
squamous papillary tumor group, 11% in the adamantinous group with a mean
follow up period of 7.5 years. The squamous papillary tumors were more
benign with a good outcome in 84.6%, and poor in 15.4%, without
mortality. Adamantous tumors fared worse with a good outcome in 51.6% in
adults and 73.9 % in children, mortality of 16.2% in adults and 6.5% in
children. In Hoffman’s review (1992) of 50 pediatric cases,
radical excision was possible in 90%, with a mortality rate of 6%. The
recurrence rate among these children was 34% with a mean follow up period
of five years. Symon (1991) reported 50 mostly adult cases of
totally removed craniopharyngiomas with a mortality rate of 4% and major
morbidity rate of 15%. The recurrence rate in his series was 6% within
the mean follow up period of 30 months.
These authors and their
results suggest that an attempt at total removal imposes no greater
burden upon these patients as a result of neural or endocrine damage than
other therapeutic modes would do.
Subtotal removal with
radiotherapy:
Most widely practiced.
When the goal of total
surgical removal is not reached, radiation therapy has the power both to
improve the survival and to prolong the interval preceding recurrence.
Radiation seems to be the best method of gaining control of tumor left in
situ.
Proponents of primary
subtotal or partial removal of craniopharyngiomas with radiation therapy
believe that their results are superior in outcome to those of radical
surgery with significantly less morbidity and mortality.
On the other hand, radiation
therapy just retards the regrowth of craniopharyngiomas, but is rarely
curative, and can lead to further endocrinological visual and
psychosocial compromise, especially to the immature brain. It does not
reverse most of the pre-treatment deficiencies, and carries the risk of
development of radiation-induced necrosis and neoplasms such as gliomas
and sarcomas.
Radiation-related morbidity
correlates strongly with the given radiation dose. In Regine’s
report (1993), 44% of all patients receiving tumor doses of < 54 Gy
developed recurrence after this treatment, while only 16% of those
receiving more than 54 Gy. However, any possible benefit in tumor control
with doses above 60 Gy at 1.8Gy per fraction appears to be offset by the
increased risk of radiation injury.
Those patients in whom
removal is deemed to be total should not be irradiated. Because of the
long-term effects of radiotherapy, some workers do not administer it
unless there is a symptomatic recurrence. Because the risk of further
endocrine and psychological injury by irradiation is related to the age
of the patient, it may be advisable to delay radiation therapy in
children for as long as possible.
In Wens’ series
(1992) of 34 cases, total excision resulted in 20% recurrence rate,
subtotal removal in 60%, and subtotal resection with radiation in 12.5%
recurrence rate within a mean follow up of 6.4 years. Wen concludes that
subtotal removal with radiotherapy is a significantly better mode of
treatment.
In Fischer’s series
(1990), management of childhood craniopharyngiomas was weighed toward
subtotal operations with irradiation. His series mortality of 8% and the
recurrence rate of 14%, as well as the social quality of life, are
comparable to or even better than the results of the more radically
oriented series.
Carmel
reported (1982) the 10 year survival rate to be 52% with subtotal removal
alone and 87% with subtotal surgery plus irradiation. Tumors recurred
within 10 years in 50% of those presumed to have had total removal, in
more than 90% of those subtotally removed and in less than 25% of those
after subtotal removal and irradiation.
Unfortunately there have
been no prospective studies comparing the efficacy of various treatment
modalities in craniopharyngioma.
Other modalities:
Instillation of
radioactive substances into the cyst isotopes such
as yttrium-90 or phosphorus-32 into craniopharyngioma cysts using
sterotaxy can successfully improve the clinical condition and survival.
About 60% of craniopharyngiomas are mainly cystic, and this rate is
higher in cases of recurrence. Injections of colloidal these implanted
isotopes directly destroy the epithelial lining of the cysts, and cause
accumulation of collagen fibres, hyaline degeneration and vascular occlusion,
thus indirectly damaging the secreting tumor cells. Follow up diagnostic
studies show gradual cyst regression, which commences a few months after
installation, with cyst obliteration in 75% of the cases. A precise
knowledge of the cyst volume is necessary to determine the amount and
dosage of radioactive isotope to be injected. The optimum dosage of
radionuclide should be sufficient to destroy cyst epithelium while
minimizing damage to surrounding structures.
Intracavitary brachytherapy
can be combined with external fractionated radiation therapy to address
the solid components of the tumor not treated by the beta-radiation
emitter. Like surgery, visual improvement is likely in patients with
moderate deficit, but is less evident in patients with severe visual
loss, while endocrine deficiencies seldom change.
Cyst drainage
(Percautaneous) drainage of a unilocular cyst by means of a
sterotactically implanted catheter, linked to a subgaleal reservoir
allows periodic aspiration of the cyst fluid. This technique also allows
the placement of radioactive or chemotherapeutic substances into the
cystic tumor in temporary or permanently inoperable cases this modality
seems to offer a safe alternative.
Radiosurgery
should be considered in cases when the solid component of primary or
residual tumor is smaller than 2.5 cm in the longest diameter. It is a
primary treatment alternative for elderly or medically infirm patients,
or for those who refuse surgery. Further follow-up is necessary to
evaluate the long-term tumor control rate and the tolerance of
surrounding critical structures.
Chemotherapy: Intracavitary
installation of bleomycin and methotrexate has been reported with
promising results. These agents might work by decreasing the secretion of
cystic fluid and causing tumor cell degeneration. Systemic chemotherapy
of craniopharyngiomas has been used sporadically in patients who refused
other methods, with some merit.
These results warrant
further evaluation of the effect of chemotherapy on craniopharyngiomas.
|
