|
Cranial
dysraphism includes a wide variety of cystic lesions, commonly known as,
‘encephaloceles’, and other midline lesions of the skull that are not
cystic, such as, rudimentary encephaloceles, congenital defects of the
scalp or skull, and congenital dermal sinuses. Neuroembryology,
pathogenesis and prenatal diagnosis are discussed elsewhere.
Encephaloceles:
An
encephalocele is a cystic congenital malformation in which central
nervous system structures, in communication with cerebrospinal fluid
pathways, herniate through a defect in the cranium. It is the result of
failure of the surface ectoderm to separate from the neuroectoderm. This
results in a bony defect in the skull table, which allows herniation of
the meninges(meningocoele) or herniation of brain tissue in addition(a meningoencephalocele).
Unlike spinal dysraphism, encephaloceles form after neural tube closure.
Those
lesions without a cystic component, such as congenital absence of the
scalp or skull, could occur at an even later date.
Incidence:
When
compared to the spinal dysraphism, they are one tenth less common.
Posterior location is common in the west, and anterior location in south
asia. In India, the occipital encephalocoele is certainly the commoner of
the two. The reason for the geographical differences is unknown.
Most
cases of encephalocele are sporadic, with only a few patients having a
positive family history for neural tube defects involving either the
cranium or the spine. Posterior lesions are more common in females.
The
role of other factors, such as nutrition, folic acid, and increased body
temperature of the mother, are not known because the rarity of this
lesion makes it difficult to obtain the appropriate epidemiological
information.
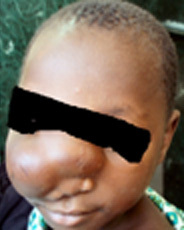
Anterior
encephaloceles can be classified into frontal, sincipital (those located
about the midface- nasoethrnoidal, nasal, nasoorbital, nasopharyngeal,
buccal), and basal (those located at the base of the skull). Almost all
of these lesions contain neural tissue and thus are meningoencephaloceles.
The neurological outcome with most anterior lesions is normal and
hydrocephalus is unusual, but the associated craniofacial disruption can
vary from minimal to massive which include hypertelorism, orbital
dystopia, elongation of the nose and other stigmata of midline or lateral
facial clefts.
The
most common location is near the midline posteriorly, in the occipital or
suboccipital region, their size, whether there is neural tissue, and how
much neural tissue is contained within the lesion have overwhelming
bearing on outcome. Anterior and middle fossae are frequently deformed
and a Chiari malformation is a frequent association. Torsion of the
spinal cord and brainstem, asymmetry of the hemispheres, ventricular
anomalies and fusion of the thalami may occur secondary to herniation of
the brain into the encephalocoele.
Cranial
meningococles may rarely occur in the temporal region as a result of a
defect in the development of the first branchial arch.
Clinical
Features:
Most
often, they present as a midline cystic mass of variable size and shape,
and usually translucent, with variable amounts of solid tissue. It may be
reducible and exhibits a cough impulse.
A
basal lesion may protrude through the superior orbital fissure causing
proptosis or may herniate through the cribriform plate or sphenoid sinus
into the nasal cavity or nasopharynx, causing nasal obstruction.
Occasionally they rupture leading to CSF leak and recurrent meningitis.
Other
associated brain abnormalities that may occur in isolation or as part of
genetic or nongenetic syndromes include spina bifida, agenesis of the
corpus callosum, Arnold-Chiari II malformation, Dandy-Walker
malformation, and brain migrational anomalies.
Neurological
deficits are uncommon. However, during follow up patients with occipital
encephalocoele may have ataxia, nystagmus, blindness, pyramidal tract
signs and mental retardation either alone or in combination. Associated
anomalies include cleft palate and congenital heart lesions.
Imaging:
|
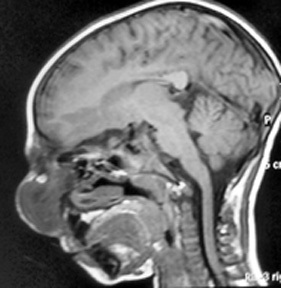
The
diagnostic study of choice is magnetic resonance imaging, supplemented
by magnetic resonance angiography if the vascularity and its relation
to the lesion need to be better defined.
With
sincipital and basal encephaloceles, computed tomography studies,
sometimes including three-dimensional reconstruction, may be of aid in
determining the need for, and planning the extent of, craniofacial
reconstruction.
Plain
radiographs are of limited value and are rarely obtained. After repair of
a posterior encephalocele, cranial ultrasound is an effective way of
following the size of the ventricles for development of progressive
|
|
|
|
|
Fronto-ethmoidal-sphenoidal encephalocele
|
Fronto-ethmoidal-sphenoidal encephalocele -MRI
(sagittal)
|
|
hydrocephalus,
which requires the placement of a shunt for diversion of cerebrospinal
fluid.
Treatment:
The
severity of additional anomalies must be considered as well as the size
of the lesions, amount of neural tissue within the sac, and degree of microcephaly,
and surgery may be withheld in selected cases.
Surgical
repair of the encephalocele involves, dissection of the sac and isolation
of the neck, inspection of the contents and replacement if possible,
excision at the neck and adequate closure of the defect at its neck.
Lesions
that are located above the torcular most often contain dysplastic neural
tissue, the resection of which does not influence neurological outcome.
Rarely, the cerebral tissue appears normal, in which case every effort should
be made to preserve it intact. This may require removal of the
surrounding bone so as to expand the cranial vault to accommodate the
volume of the neural tissue. Lesions below the torcular pose problems
similar to those found above, with the additional possibility of
inclusion of brain stem and cerebellum in the encephalocele. Large
lesions can be associated with a significant arterial supply and venous
drainage. The vessels therefore must be identified and coagulated before
being divided, and the sagittal, transverse, and occipital sinuses as
well as the torcular must be located before dural resection.
Intracranial
repair with craniofacial reconstruction is preferred in most sincipital
encephaloceles.
The
operative approach to a basal encephalocele is similar to that for the
sincipital type. Basal encephaloceles differ from sincipital
encephaloceles in that the bony defect is more posteriorly located on the
cranial base, especially involving the sphenoid. They are more likely to
contain structures such as the hypothalamus, pituitary gland and stalk,
optic nerves, optic chiasm, and anterior cerebral arteries within the
herniation. In infants, the presence of a basal encephalocele may be
heralded by nasal obstruction. As the mass presents in the nose, it may
be mistaken for a nasal polyp, the biopsy of which can lead to
cerebrospinal fluid rhinorrhea.
The
differentiation between a polyp and encephalocele is that an
encephalocele pulsates, presents medially from the nasal septum, and
widens the nasal bridge, while a polyp does not pulsate, is located
laterally, emanates from the turbinates, and does not widen the nasion.
Any
associated hydrocephalus is treated by a shunt procedure before the
encephalocoeie is excised. Following a shunt the encephalocoeie becomes
smaller, the closure of the defect becomes easier and the postoperative
risks of wound breakdown and CSF leak are minimized.
A
water-tight closure of the dural defect is achieved, using a graft if necessary.
A large defect in the bone may require repair.
Postoperatively
a careful watch is kept for evidence of raised pressure as a result of
hydrocephalus. Occasionally, in the immediate postoperative period the
intracranial pressure may rise very rapidly over a few hours and result
in a fatality unless recognized and treated energetically. If there is
evidence of rising intracranial pressure, the immediate postoperative
period is tided over by ventricular punctures. Some of these cases
require a shunt procedure postoperatively. Anticonvulsants are routinely
used. In occipital encephalocoeles, the outcome depends on the amount of
brain in the herniated sac, as well as the presence or absence of
hydrocephalus. A third of the patients develop normally, another third
are educable with moderate retardation and physical defect. The remaining
third are left with severe deficits. The prognosis in anterior
encephalocoeles is excellent.
Rudimentary
encephaloceles are midline lesions that are usually found at the vertex
and are associated with various skin changes, which can include cutis
aplasia, abnormal hair and hair pattern, telangiectasia, excessive
fibrous tissue, and dysplastic glial elements.
The
dysplastic skin is removed for cosmesis as well as to eliminate the local
irritation and pain that can accompany these defects. In lesions with
intracranial connection, intracranial exploration is avoided.
A
congenital dermal sinus is most likely to be found in the occipital
location, and often associated with a dermal inclusion cyst, which may
lie extradurally or, if it is intradural, may be located within the
cerebellum, fourth ventricle, or cisterna magna.
Chiari malformations are
discussed elsewhere.
Anencephaly:
Anencephaly results from failure of neural tube
closure at the cranial end of the developing embryo. Absence of the brain
and calvaria may be partial or complete. The brain is represented by a
dysfunctional mass of nervous tissue, which includes the primitive brain
stem and a few cranial nerves, which may be exposed or covered with skin.
Parts of the brain stem and spinal cord may be missing or malformed.
Failure of the neural groove to form into a tube, or gain a covering of
mesoderm or ectoderm, results in exposure of the brain and its ultimate
degeneration. Most of these fetuses are stillborn, but some may
occasionally survive for a few hours. The fetus has a partially destroyed
brain, deformed forehead, and large ears and eyes with often relatively
normal lower facial structures. Anencephaly and spina bifida are the most
common type of dysraphism. Familial occurrence can be high.
|
