|
Astrocytomas are
the most common (over 70%) of all primary intracranial neoplasms.
Spinal
astrocytomas are discussed elsewhere.
The world health
organization (WHO) currently recognizes multiple astrocytic tumor
variants.
They may be classified according to
their cytologic characteristics, viz., fibrillary, protoplasmic or
gemistocytic; or according to their location, viz., cerebral,
hypothalamic, cerebellar or brainstem. The latter classification
apart from location is also based upon considerations, partly, of cell
type and partly of growth and behavior of the tumor.
Although tumor composition is often heterogenous, fibrillary
astrocytes are by far the most frequently observed. Astrocytes
show a stellate arrangement of fine fibrillary processes. Their nuclei
are oval with scattered chromatin. Diffuse cerebral astrocytomas are
composed of cells with this appearance, and are predominantly of the fibrillary
type. These tumors may contain microcysts and foci of dystropic calcification.
Identifying the
presence of
these morphological subtypes, however, appears to have little value in terms of predicting prognosis.
A possible
exception is the 9% to 19% of astrocytomas composed
predominantly of gemistocytic cells.
In such cases, gemistocytic cell content in excess of 60% has been associated with aggressive neoplastic behavior and a poorer
prognosis. Microscopically,
the cells are large, globoid, and packed with a hyaline cytoplasm,
and eccentric nuclei with coarse chromatin and conspicuous
nucleoli. They show glial fibrillary
acidic protein (GFAP) positivity.
Protoplasmic variant, the least common
variant (<1% of all astrocytomas), involves cerebral hemisphere
superficially. Histologically homogenous population of small astrocytes
with few delicate processes.
Over the years, the
notion that histological characteristics are useful
as predictors of neoplastic aggressiveness has come to be
accepted. As a result, current classification systems
assign distinct tumor grades based on the presence, absence, and degree of specific observable histological
criteria. These criteria commonly include the overall degree of tumor cellularity, extent of cellular and
nuclear pleomorphism,
frequency of mitotic activity,
and presence or absence of necrosis and endothelial proliferation.
In the WHO four tier system, similar weight is given to the presence of nuclear atypia, mitoses, endothelial
proliferation, and necrosis. In the absence of these criteria, the grade
of 0 is assigned. If any of the criteria can be identified, a grade of 1 is aligned. Successive grades
up to 4 are assigned upon subsequent identification of any of the
remaining criteria. Using this system, the proportionate distribution of
all astrocytomas has been found to be 4.1% grade 1, 23% grade 2, 16%
grade 3, and 57% grade 4.
We follow Daumas- Duport (St.
Anne/Mayo,1988) system. This scheme is restricted to astrocytomas and
glioblastomas. The following histological abnormalities are used to place
tumors in 4 grades: nuclear atypia, mitosis, endothelial proliferation,
and necrosis. The system is very
reproducible and the grade is strongly correlated with survival.
|
WHO designation
|
WHO grade
|
St. Anne/Mayo grade
|
|
Pilocytic astrocytoma
|
I
|
excluded
|
|
Astrocytoma
|
II (nuclear
atypia and no/or rare mitosis)
|
1 (no criteria
fulfilled)
2 (one
criterion: usually nuclear atypia)
|
|
Anaplastic astrocytoma
|
III (nuclear atypia and marked mitosis)
|
3 (two criteria: usually nuclear atypia and mitosis)
|
|
Glioblastoma
|
IV (nuclear
atypia, mitosis and endothelial
vascular proliferation and necrosis).
|
4 (three or four
criteria: usually the above and necrosis and or endothelial
proliferation).
|
However, when
describing the histological grade of astrocytic tumors, communication is
often simplified by use of of the terms' low grade astrocytoma',
'anaplastic astrocytoma', and 'glioblastoma mutiforme'. Despite variability among the grading systems, the
distinction between low grade astrocytoma, and anaplastic astrocytoma is
commonly made based on the presence of mitoses and increased cellularity.
Similarly, glioblastoma multiforme and anaplastic astrocytoma are
frequently distinguished based on the
presence of necrosis and endothelial proliferation.
Individual variants of astrocytoma may display both age and
location related predilections.
For example, the
majority of pilocytic astrocytomas occurs in childhood population and
involves the cerebellum.
A tendency to the
diagnosis of higher tumor grades is found with
increasing patient age. This is reflected by the presence of peak incidences for low grade astrocytoma, anaplastic
astrocytoma, and glioblastoma multiforme during the third, fourth, and fifth decades of life,
respectively. Further, the overall incidence of astrocytomas, regardless
of grade, is seen to rise proportionately with age, peaking during the
fifth to sixth decades of life.
Clinical
features of this condition commonly include headaches,
nausea, papilledema,
and blurred vision. Symptoms such as these are of limited value for predicting tumor location, although they may
play a valuable role in prompting patients to seek medical attention. Other symptoms experienced by patients with
astrocytomas are primarily
determined by the location and size of the tumor involved. Supratentorial
lesions present with seizures and focal neurological deficits, such as,
dysphasia and hemiparesis. In
cases involving the posterior fossa,
ataxia, dysmetria, and nystagmus, are frequently found. Astrocytomas involving the brainstem are often
notable for the production of a
variety of symptoms, including cranial nerve deficits and limb
weakness. in the presence of large tumors or obstructed cerebrospinal
fluid (CSF) circulation, evidence of
increased intracranial pressure
may be seen.
In general
magnetic resonance imaging is superior to computerized tomography
to evaluate their composition and relationships with nearby anatomical
structures. Conversely, CT is more sensitive for revealing characteristics such as hemorrhage and intratumoral
calcification. Despite the excellent radiographic methods available, however, imaging studies are often unreliable for discriminating among
the individual historical
grades and variants of astrocytoma.
The use of post
operative imaging (within 48 hours), particularly MRI with gadolinium, is
invaluable is assessing completeness of resection and detecting
recurrence.
The
astrocytoma, anaplastic astrocytoma, and glioblastoma are regarded as a
spectrum of diffuse astrocytic tumors with common molecular genetic
abnormalities. The presence of multiple characteristic genetic mutations, whether
inherited or acquired, has been
associated with the oncogenesis of astrocytomas. Some have also theorized that the accumulation of specific
genetic mutations brings about the further progression of low grade neoplasms to higher degrees of
malignancy. For example,
deletion mutations of chromosome 17 (17p) and, less frequently, chromosome 22 (22q), are known to be present
among a large number of
astrocytomas regardless of grade. Other mutations 13 (13q), 9 (9p), and 19(19q), possibly reflecting later transformative events. Moreover, deletion
mutations of chromosome 10 (10q) are found almost exclusively among
glioblastomas, suggesting that they are involved in the transition to the
highest grades of malignancy.
In astrocytic
tumors, the transition to glioblastoma is associated with upregulation of
the epidermal growth factor receptor (EGFR) gene found on chromosome 7. It has been proposed that
mutation common to high grade astrocytomas
specifically that of chromosome 10q may be involved in the stimulation of
EGFR gene expression or the disinhibition of
its regulation.
Significant interest has been generated in utilizing the presence
of these characteristic genetic alterations for the purpose of predicting aggressive tumor behavior. In the future, the use of cytogenetic
techniques is likely to play an increasing role in the treatment of
neoplasms, such as the astrocytoma.
Low grade Astrocytoma:
The
term 'low grade astrocytoma� is given to a group of astrocytic tumors
with a relatively well-differentiated histological appearance. Among these are included typical low grade astrocytoma, pilocytic
astrocytoma, pleomorphic
xanthoastrocytoma, and subependvmal giant cell astrocytoma. Although strictly intended to
imply uniform astrocytic cell
composition, low grade neoplasms of mixed
oligodendroglioma-astrocytoma cell content are also occasionally included in this category.
Pilocytic and low-grade astrocytomas are encountered most frequently, accounting for 43% of astrocytomas as a whole.
The median age of
patients with low grade gliomas is approximately 35 years. There is a
biphasic age distribution, with two peak incidences at 6 to 12 years, and
26 to 46 years, with a slight male preponderance.
Most low grade astrocytomas in adults arise supratentorially; half are
of typical histology, with the remaining consisting
of pilocytic astrocytomas and mixed oligodendrogliomas-astrocytomas
in nearly equal proportions.
Involvement of the
hemispheres is more common than that of deeper
structures, such as the basal ganglia and
brainstem.
When hemispheric
in location, frontal lobe involvement is more prevalent than temporal or
parietal lobe involvement.
Infratentorial
lowgrade astrocytomas occur most commonly in the
cerebellum of children. Whereas low grade cerebellar astrocytomas account
for 15% to 18% of all intracranial tumors among children, they
account for only 1% of those among adults. The pattern of cerebellar
involvement is unihemispheric in approximately 30%, bihemispheric in 34%, and vermian in 16% of cases. Histologically, the vast majority, approximately 85% of lowgrade cerebellar astrocytomas are pilocytic.
For practical purposes, pilocytic
astrocytomas are commonly given separate consideration from
typical low grade astrocytomas as they
have been associated with a more favorable
prognosis. In addition to the cerebellum, pilocytic
astrocytomas (juvenile type) commonly involve the hypothalamus and optic pathways, constituting the majority of tumors
referred to as hypothalamic
and optic gliomas.
Pilocytic astrocytomas
diagnosed during the first two decades of life are predominantly cerebellar. In contrast,
those found in adults are most
often supratentorial. When located supratentorially, the temporal
lobes, frontal lobes, and basal
ganglia are most commonly involved. Pilocytic astrocytomas are typically diagnosed earlier in
life than low grade
astrocytomas, with mean ages ranging
from 14 to 18 years, and from 30 to 37 years, respectively.
Signs and symptoms observed at the time of diagnosis are
primarily related to tumor location. 60% of them present with seizures,
twice as frequently as the high grade ones. They are one of the common
causes of intractable epilepsy.
|
The CT
characteristics of typical low-grade astrocytomas are those of a poorly defined, hypodense mass. Unlike high grade astrocytomas, less evidence of mass effect,
surrounding edema, and heterogeneity is present.
On MRI,
these tumors are hypo to isodense on T1 and hyperdense
on T2 images. Enhancement is variable or absent on both CT and MRI. Calcification and cytic
changes are not rare. In comparison, pilocytic
astrocytomas have clearly defined borders and are further distinguished by their tendency to enhance,
faintly and heterogenously.
|
|
|
|
|
|
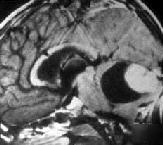
Cystic
Pilocytic astrocytoma-MRI
(with
mural nodule)
|
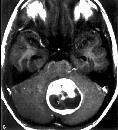
Pilocytic
astrocytoma-MRI
(well defined
borders
and
no edema)
|
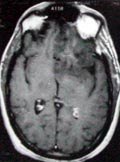
Low
grade astrocytoma- MRI
(ill
defined borders)
|
|
Other recent advances in neuroimaging
help for further evaluation.
|
|
|
|
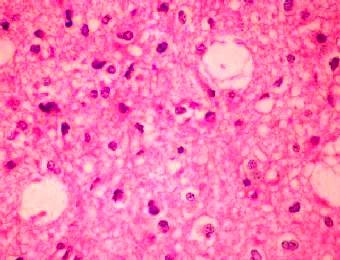
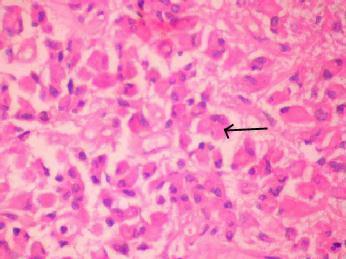
Fibrillary
Astrocytoma (H&E)- moderate increase in cellularity by
neoplastic astrocytes with enlarged nuclei and coarse chromatin.
Cytoplasmic processes
are
indistinct in a finely fibrillary background.
|
Gemistocytic
Astrocytoma (H&E)- distinct cells with large
eosinophilic, slightly angulated (arrow)
cytoplasm
and eccentric nuclei.
|
On gross
examination, low grade astrocytomas are typically poorly circumscribed
and may be similar in appearance to the surrounding non neoplastic
tissues. On histological examination, mildly
increased cellularity and slight pleomorphism are
present, but notably absent are changes such as mitosis and extensive
atypia, which are characteristic of high-grade astrocytomas. On
microscopic examination, fibrillary astrocytes are most
often observed, though multiple astrocytic morphologies
may also be present. Neoplastic cells can be seen to diffusely infiltrate surrounding tissues. Nonneoplastic
tissue elements may become incorporated, or trapped within the tumor
mass. This phenomenon may on occasion give false impression of mixed
tumor.
|
Pilocytic astrocytomas are composed of a
biphasic cellular pattern consisting of bipolar
"piloid" cells with multiple, long
fibrillary processes and microcystic structures made up of sparsely
fibrillated proto�plasmic
astrocytes. Also characteristic of pilocytic astrocytomas is the presence of eosinophilic
structures, seen as intracytoplasmic globules or as long extracellular
fibers. These structures have been termed �granular bodies� and
�Rosenthal fibers� respectively. Evidence
of malignant changes, such as atypia and endothelial proliferation,
can
frequently be seen. Unlike the situation for typical low grade
astrocytomas, the presence of these changes does not necessarily
predict aggressive neoplastic behavior. Key features include
Rosenthal fibers and/or
eosinophilic hyaline granules.
The management of low
grade gliomas continues to be controversial. All treatment options,
including radiotherapy immediately after surgery, radiotherapy only for
incomplete resected gliomas, and radiotherapy at recurrence or
progression, are considered valid options, and none is supported by a
randomized controlled study.
|
|
|
|
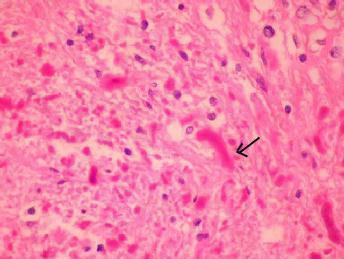
Pilocytic
Astrocytoma(H&E)-Aggregates of Rosenthal fibres(arrow)
and granular eosinophilic bodies are seen along
with
fibrillary fasicles.
|
|
Many
consider a complete resection to be curative for pilocytic astrocytomas with the use of
postoperative irradiation offering little additional benefit.
furthermore, some have advocated withholding radiation treatment, even if
subtotal resection is obtained initially, reserving radiation for tumor
recurrence or surgery limited biopsy. The timing and extent of
resection is controversial. A similar argument has been made for the treatment of typical low grade
astrocytomas in both children
and adults.
Restrictions on
the use of radiation treatment among young children
have been recommended due to high degree of associated morbidity. Additionally,
some have implicated radiation as a cause of the mutational events
leading to increased aggressiveness. Despite this, some have found post
operative radiation useful in
typical low-grade astrocytomas, even in cases of total resection. Still others have argued that the
benefit of completely resecting typical low grade astrocytomas remains unproven, instead advocating the use of
focused radiation therapy alone as initial treatment.
The use of chemotherapeutic agents for the treatment of low grade astrocytomas is controversial, with studios citing
little improvement over the results achieved with conventional therapies
alone.
These studies have
recommended reserving chemotherapeutic agents for cases of inoperable low
grade astrocytomas or as a means of obviating
the morbidity associated with cerebral irradiation in children.
The prognosis for
patients with low grade astrocytomas varies with tumor location and
histology.
Regardless of
supra or infratentorial location, pilocytic tumors are associated with the most favorable prognosis, with 5 and 20 year survival rates of 85% to 86% and 79% to 82%.
Although varying
with supra and infratentorial location, the
prognosis for other
low grade astrocytomas is much less favorable.
Those in a supratentorial location have
been found to carry 5 and 10 year
survival rate of 51% to 56% and 23% to 39% respectively. In contrast, those occurring in the
cerebellum are associated with still poorer outcomes, with survival rates
at both 5 and 10 years of 7%.
Histologically, the microcystic
change is recognized to be a regressive feature and indeed one does not
witness much cellular activity in such regions. The cytoplasm is scanty
and fibrillar and the nuclei small and monomorphic. Another feature
of slow growth and thus seen in low grade astrocytomas is the formation
of thick smooth cytoplasmic extensions of glial cells, called Rosenthal
bodies. They were believed to represent degenerating astrocytes.
Other factors
predictive of improved outcome include younger age, seizure at
presentation, and lack of preoperative fixed neurological deficit.
Tumor recurrence is often associated with malignant progression and is a common cause of mortality among patients
with low grade astrocytoma. The frequency is thought to be highest among patients with typical low grade astrocytoma, occurring in 57% to 72% of cases. Factors that have been associated with an increased
rate of an increased rate of recurrence include subtotal resection and
the presence of oligodendroglial tumor components. additionally, some
believe that malignant progression of low grade astrocytomas is more
prevalent among adults.
Research efforts for the low
grade astrocytomas focus on developing chemotherapy regimens that control
tumor growth with fewer side effects on other organs of the body. Because
these tumors grow slowly, the strategy is to give less intensive
chemotherapy over long periods of time.
For older children and those whose
tumors progress despite chemotherapy, new radiation techniques are under
study to �focally� deliver therapy with minimal effects on the normal
brain.
Subependymal
giant cell astrocytoma (SEGA): discussed elsewhere.
Pleomorphic
Xanthoastrocytoma (PXA):
They are
rare(<1%), typically, develops in children and young adults.
Invasion of the
overlying dura in superficial lesions is common. Occasionally,
skull may be
eroded. Temporal lobe is the commonest site, followed by the parietal,
occipital, and the frontal lobes.
|
A history
of chronic seizures and headaches is the usual presentation.
On
MRI, T1 images reveal, an iso to hypodense lesion with cystic and
calcified changes. Uniform contrast enhancement of the tumor nodule
with typically non enhancing cyst wall is seen. On T2 it is hyperdense.
Gross
total excision, if possible, is advised. The cyst wall need not be
removed. The role of adjuvant therapy following a subtotal resection or
in those with high mitotic index is not clear at present.
|
|
|
|
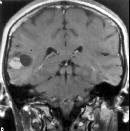
PXA-MRI
|
|
|
Resurgery
for recurrent or progressive lesions followed by radiation is
recommended.
Histologically, there is closely packed, highly pleomorphic, giant and
multinucleated cells. Variable xanthomatous change is seen in the
cytoplasm. Prominent eosinophilic granular bodies are constant. Mitosis
is rare. In children, it may mimic a GBM.
Pre
immunohistochemistry days, it was classified as histiocytic fibromas.
Some still call it gliofibroma and group this along with gangliogliomas
and infantile desmoplastic gliomas.
The
astrocytic nature is demonstrated by GFAP immunopositivity.
The
outcome is generally good. Local recurrence may occur.
15% of
the cases recur and undergo malignant change into GBM.
Anaplastic Astrocytomas(AA):
|
|
|
|
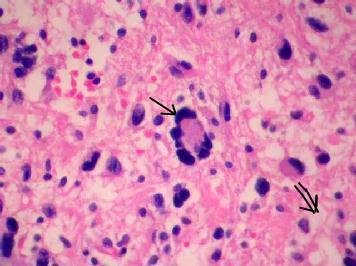
PXA
(H&E) -
Fibrillary
and giant often multinucleated neoplastic astrocytes (arrow) intermingled
with spindle cells, and
xanthomatous
cytoplam(double arrow).
|
|
|
Anaplastic
astrocytomas account for approximately 12% to 34% of high grade (WHO Grade III)
astrocytomas. Their peak incidence occurs during the fourth to early fifth
decades of life, falling between that of low grade astrocytomas and
glioblastoma multiforme. They
are also intermediate among
astrocytomas with respect to histology. While distinguishable from low
grade astrocytomas on the basis of their increased cellular density,
greater degree of nuclear atypia, and mitoses, they lack the
endothelial proliferation and necrosis characteristic of glioblastoma
multiforme.
Radiological imaging reveals better defined borders than that of low grade
ones; they appear hypo to iso dense on T1 and hyperdense on T2 MRI
images. Greater the contrast enhancement, and edema suggest a higher
grade, and unlike glioblastoma, the enhancement is homogenous.
Anaplastic astrocytomas are particularly
susceptible to histological misclassification, often being
diagnosed as glioblastomas. Additionally confusing is the finding that
low grade astrocytomas with a gemistocytic astrocyte content in excess
of 60% behave with an aggressiveness similar to that of anaplastic
astrocytomas and often treated as anaplastic.
Grossly, anaplastic astrocytomas have a more circumscribed
appearance than the low grade
astrocytic tumors, but friable, granular, and grayish. They are
prone to hemorrhage. However, this
appearance is deceptive as
neoplastic elements can still be found to infiltrate surrounding
tissues. Microscopically, neoplastic cells can be variably small,
large, stellate, pilocytic, etc.
Key feature is usually mitotic
figures.
|
|
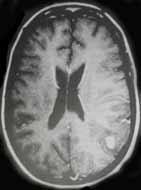
|
|
Anaplastic
astrocytoma-MRI
(more
homogeneous contrast enhancement than GBM)
|
|
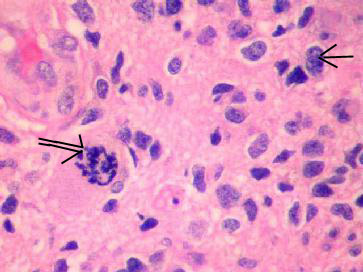
|
|
Anaplastic Astrocytoma (H&E)-moderate to
marked increase in neoplastic cellularity(arrow),
cellular pleomorphism, & mitosis(double arrow).
|
|
The median
survival for patients diagnosed with anaplaslic
astrocytoma ranges from 15 to 28 months, with projected 1, 2, and 5 year
survival rates of 60% to 80%, 38% to 64%, and 35% to 46%, respectively.
Aggressive resection has shown higher survival; some have found little
improvement with radical excision and place a greater emphasis on the
radiotherapy and chemotherapy.
The use
of postoperative radiation has been shown to prolong survival in
patients with anaplastic astrocytomas, often to a greater degree than
when used for the treatment of
glioblastoma multiforme. In addition,
some authors have advocated the use of alternate treatments, such as brachytherapy,
radiosensitizers, and chemotherapeutic agents both initially and
at recurrence. However, others have
found these alternate modalities to be of little extra benefit and
rely more heavily on conventional forms of treatment.
For high grade tumors, new approaches on
trial, include use of new chemotherapy drugs, high doses of
chemotherapy following radiation therapy, and gene therapy to make the
tumor cells more sensitive to chemotherapy. A major problem in treatment
is that the high dose chemotherapy also kills cells in the bone marrow
that produce healthy blood. This raises the risk of severe infection and
slows down the delivery of chemotherapy. Gene therapy approaches are
being developed to protect bone marrow from these side effects so that
chemotherapy can be given more intensively to fight the rapid tumor
growth.
Genetherapy
and immunotherapy are still under in the experimental stage.
As with other
astrocytic tumors, primary site recurrence is the
most common cause of mortality.
Factors thought to correlate with improved outcome in patients with anaplastic astrocytoma include younger age,
higher preoperative performance scores, and presentation with seizures.
Additionally, some have found improved outcomes among
patients previously diagnosed with lower grade
astrocytic tumors in comparison to patients in whom an
anaplastic astrocytoma has arisen de novo.
Glioblastoma
multiforme (GBM):
First recognized by Virchow, in
1863, and described later as 'spongioblastoma multiforme', this, the most
malignant neoplasm in the human body. Glioblastoma
multiforme is the least differentiated and most aggressive form of
astrocytoma.
It accounts for
15% to 23% of all primary intracranial tumors.
Furthermore, it
constitutes 35% of gliomas, 66% to 87% of high grade astrocytomas, and
50% of all astrocytomas, making it the most common astrocytoma.
Patients diagnosed
with glioblastoma multiforme are most commonly in their fifth or sixth
decade of life.
The diagnosis is
made less frequently in younger age groups and rarely in children, where
GBM account for less than 9% of all
|
intracranial
primary tumors.
Regardless
of age, hemispheric location is most common.
The
presence of multifocal tumors is thought to occur in 2/3% to 9% of
cases.
Headaches due to raised ICT,
and focal neurological deficits according to the site of location are
the common presenting symptoms. Unlike in low grade gliomas,
seizure as a presenting symptom is uncommon.
On CT
and MRI, the GBM appears as a well defined mass with
heterogenous contrast enhancement and extensive parenchymal edema. A
characteristic irregular rim of high intensity (due to florid
endothelial/vascular proliferation), may simulate metastasis or an
abscess.
|
|
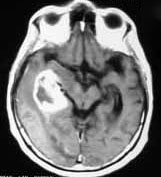
|
|
Glioblastoma-MRI
(with
rim of heperdensity and edema)
|
|
Most GBMs contain
a centrally located, hypoxic area of necrosis that develops as the tumor
mass outgrows its blood supply. This hypoxic zone is concentrically
enveloped by hypercellular neoplastic tissue and surrounding edematous
white matter. The
more malignant astrocytomas have been found to have features
histologically indistinguishable from glioblastoma multiforme and thus
there is a a controversy against assigning a separate name for this
tumor, as there is no such cell as a glioblast.
|
However, it must be admitted
that a percentage of glial tumors present themselves with such a very
rapid onset of signs and symptoms, that either at surgery or autopsy
there is no clear trace of an astrocytoma and all parts of the tumor
show merely the characteristic pleomorphism of a glioblastoma.
Microscopically,
the tumor is highly cellular with closely packed cells exhibiting a
varying degree of pleomorphism. The cells vary in size and shape; large
bizarre giant cells with many nuclei are frequently seen, as also
hyperchromatism, mitotic figures and abnormal nuclei. Another
striking feature is the presence of vast areas of necrosis ringed
closely by growing spongioblasts giving rise to an appearance of
pseudopalisading. Mononuclear cuffing of blood vessels and
endothelial proliferation, constitute further histological evidence of
a higher degree of malignancy. The malignant astrocytomas, in
particular, tend to spread along the meninges after reaching the
surface, and along the blood vessels after entering the Virchow-Robin
spaces.
|
|
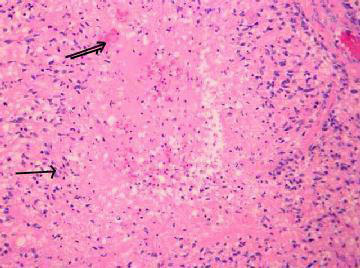
|
|
Glioblastoma
multiforme- Pseudo palisading(arrow)
of tumor cells around a central zone of necrosis(double arrow)
|
|
WHO currently
recognizes two histological variants of glioblastoma:
|
giant cell
glioblasloma in which a predominance of multinucleated giant cells is
seen, and gliosarcoma (a term originally used by Stroebe, in 1895) or Feign tumor, where malignant
neoplastic induction of vascular stromal elements is present. An
invasive mesodermal tumor from either the meninges of the blood
vessels was believed to stimulate a vigorous proliferative
and hyperplastic reaction of the surrounding neuroglia
which acquire malignant features. Protagnonists of this latter theory
are few and it is generally thought that the pronounced vascular
proliferation is responsible for a gliosarcoma. These tumors
macroscopically may sometimes resemble meningioma and even
histologically a diagnosis of mesenchymal tumor may be made if the
sampling of the tissue is not representative. Because of the
presence of mesenchymal elements, extracranial metastasis is possible
from such neoplasms.
Management, currently recommended, is an aggressive surgical resection,
if possible, and post operative irradiation as the initial form of
treatment. Attempts to achieve an
aggressive resection may, however, be limited when patients are poor
surgical
|
|
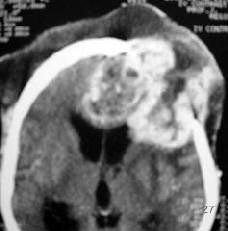
|
|
Gliosarcoma
(with
subcutaneous extension)
|
|
candidates or have
tumors that involve eloquent or deep structures. Alternate modalities are
needed in such cases.
Reoperation for
recurrence in selected patients who have had favorable results to initial
treatment may be considered.
Postoperative
radiation has been found to be helpful. Radiotherapy, age, and performance status have
been demonstrated to be the three most significant prognostic factors. Usual total dose
is 60 Gy. Newer techniques, such as,
dose fractionation, stereotactic radiotherapy, heavy particle
radiotherapy, and brachytherapy have also been used with no evidence to
suggest a better outcome.
As compared to
radiotherapy, role of chemotherapy
is limited. However, it is currently used in the young and in recurrence
after radiotherapy. �Standard�
chemotherapy has been a nitrosourea based regimen. Newer promising
chemotherapy includes Temozolomide and CPT-11.
Newer therapies,
such as, genetherapy
and immunotherapy are
under trial.
Prognosis for patients with glioblastomas have shown little
improvement despite the use of multiple treatment modalities, including
surgery, whole brain, local, and focused radiation(
brachytherapy); radiosensitizing agents, and other
forms of chemotherapy have not helped. Median life
expectancies of 8 to 10 months after diagnosis are common, along with 1, 2, and 5-year survival rates of 30% to 44%, 10% to 12%, and 2.5% to 5% respectively.
Survival rates
cited for children are similar to those for adults.
The most common cause of mortality among patients with glioblastoma
regardless of age is recurrence.
Younger patients
with seizures at presentation, lack of focal neurological deficit, and
complete tumor resection favor a better prognosis.
|
Multifocality:
When using the term "complete resection," the propensity
for astrocytomas to disseminate must be taken into account. Microscopically, local
dissemination is reflected
by the presence of neoplastic cells that often infiltrate 1 to 3 cm
into adjacent tissues despite a well-circumscribed appearance upon gross inspection. In addition, more extensive spread
occurs preferentially along sub cortical white mater tracts
(corpus callosum, uncinate fasciculus, auditory and visual bundles,
corona radiata, subependymal route, CSF dissemination, along
blood vessels & perivascular spaces, and sub pial spread frequently
giving rise to nearby tumor
foci.
Contralaterai
hemispheric spread may therefore take place by virtue of
extension through corpus callosum giving rise to characteristic
�butterfly� pattern seen on CT/MRI axial sections.
Multifocal
gliomas can be categorized as 'Connected (microscopic parenchymal
connection or
|
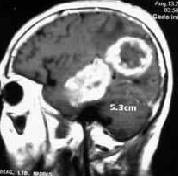
|
|
|
Multiple glioma-MRI
|
|
satellite
lesions) or Disconnected (no detectable microscopic connection)', and as
'Synchronous (if present on initial presentation) or Metachronous (if
developed during follow-up. They are termed multiple, if present at the
same time but are separate spatially, and multicentric, if they are
independent spatially as well as temporarily.
Although multiple astrocytomas may arise independently within a
single patient, the majority are probably represent the presence of a
single neoplastic disease.
Reportedly, the
multifocality occurs in 2.3% to 9.1% of cases.
Anaplastic astrocytoma has more
infiltrative growth than does GBM thus multifocal glioma occur more
frequently in AA than GBM.
Proteins
responsible for tumor cell attachment & migration are - myelin, ECM
protein, Merosin, fibronectin, laminin.
Leptomeningeal
gliomatosis:
Diffuse
subarachnoid dissemination of intracranial tumors is termed
leptomeningeal gliomatosis. This condition often results from the
presence of a high grade intracranial neoplasm that has gained access to
the CSF by virtue of its proximity to the ventricles
or cisterns. In such cases, patients may experience a
variety of
symptoms, including mental status changes, headache, cranial nerve deficits, and back pain.
Diagnosis is by CSF cytology. Radiology
may be negative.
Therapeutic
measures employed include craniospinal radiation, and systemic or
intrathecal chemotherapy. Survival rates are generally poor and primarily
related to histology of the and its responsiveness to treatment. With respect to astrocytic tumors, limited success has been achieved in the treatment of leptomeningeal
gliomatosis involving anaplastic astrocytomas.
However, the
prognosis for patients with leptomeningeal gliomatosis resulting from GBM
is bleak, with survival rates generally measured in
terms of weeks.
Extraneural
metastasis:
Among adults,
astrocytomas have the distinction of being the
intracranial neoplasm most likely to metastasize outside the CNS.
However, even with astrocytomas, metastasis is rare. The extraneural
presence of metastases is frequently associated with previous craniotomy
or a diversionary shunting procedure. It is by virtue of these routes the
metastatic cells are believed to gain access to extradural
lymphatic and vascular tissues. However, a prior dural disruption is not
strict requirement. These cases are a result of invasion of intracranial
vascular structures, such as venous sinuses.
The most common
sites for extraneural metastases include lung, lymph nodes, and bone.
In those who have
had shunt procedures done, the abdomen should also be considered a
potential site.
The probablity of
metastases appears to be related to the degree of tumor anaplasia, with
GBM more likely to metastasize than others. Survival rates for such
patients are poor, ranging from 6 months to 2 years after the time of diagnosis. Chemotherapy, although of
minimal benefit to survival, is advised to improve the quality of life.
|
