|
They are predominantly intraventricular
and account for 1%-3% of all primary brain tumors.
In children, they constitute 10% of
intracranial tumors and are third in frequency after astrocytoma and
medulloblastoma.
Some authors have found a predominance
in the first two years of life. However, congenital ependymomas are
very rare and they are often malignant, and in most cases, located above
the tentorium.
A second incidence peak is found between
the third and fifth decade.
Pathology:
Since
Bailey and Cushing’s in 1926, a lot of classification and grading systems
were proposed.
WHO
classifies ependymomas as follows:
Ependymoma (grade II) - Cellular, Papillary, Clear cell, and Tanycytic
Anaplastic ependymoma (grade III)
Myxopapillary ependymoma (grade I)
Subependymoma (grade I)
Ependymoblastomas,
are
rare and malignant, with distinct ependymal differentiation, and included
in the group of embryonal
tumors.
Intracranial
lesions are 1) Ependymomas, 2) Anaplastic ependymomas, and 3)
Subependymomas.
Myxopapillary
ependymomas are almost exclusively located in the region of the cauda
equina and originate from the filum terminale.
Ependymomas are thought to arise from
the ependymal epithelium of the ventricles and central canal. In
children, posterior fossa is the most common site, whereas it is equally
distributed through out the CNS in adults. Floor of the fourth ventricle
is the most common intraventricular location, followed by the
lateral and third ventricles. Tumors which originate at the floor of the
fourth ventricle may grow through the foramen of Magnendie into the
cisterna magna and furthermore, through the foramen magnum into the upper
cervical spinal canal as far down as the level of C5. Ependymomas
which arise from the medullary velum at the lateral recess may extend
through the foramina of Luschka into the cerebellopontine cisterns.
|
Purely
extraventricular ependymomas originate, probably as a consequence of
embryonal disturbance in the folding of the neural tube, from nests of
ependymal cells incorporated into the parenchyma of the cerebral
hemispheres.
Hemispheric
ependymomas are often cystic and malignant, with increasing
malignancy the further from the ventricle. These anaplastic
tumors arise most frequently in children, whereas the typical
ependymomas at the foramen of Monro usually occurs in juveniles
and is characterized by an absence of cellular pleomorphism and
mitoses.
Histologically,
this tumor is distinct from the central neurocytoma of the same
location.
Ependymomas
of the septum pellucidum may extend into both lateral ventricles, the
third ventricle, and the aqueduct. Ependymomas of the sella
turcica arise from the infundibulum or ependymal cell nests in the
posterior pituitary.
In
the spinal cord,
they occur in the second to fourth decades. Spinal ependymomas are
intramedullary and often found in the lumbosacral region. At
times, they extrude from the conus medullaris and remain suspended
amongst the roots of the cauda equina. They are characterized
histologically by a myxopapillary appearance. These
tumors are particularly benign, made up of small monomorphic cells, and
have a better prognosis with longer postoperative survival than
ependymomas of the brain.
Grossly,
ependymomas are firm, pale to reddish in color, nodular, partially well
demarcated, and occasionally lobulated. Cellular, epithelial, and
papillary variants have been described.
|
|
|
|
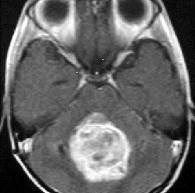
4th ventricular
ependymoma-MRI
|
|
|
|
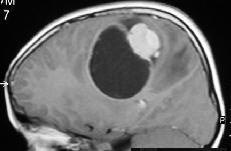
Parietal ependymoma-MRI
|
|
|
|
|
|
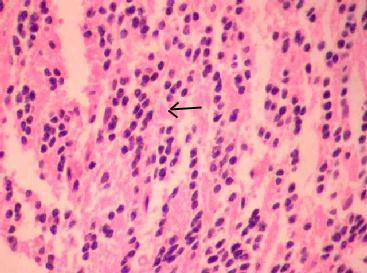
Ependymoma (H&E)-
characteristic pseudo rosettes and vascular elements surrounded by an
acellular zone(arrow)
|
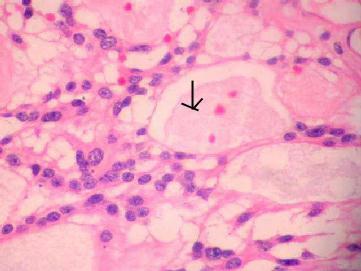
Myxopappilary
ependymoma (H&E)-Perivascular orientation of cells
forming pseudo rosettes, rare true rosettes and
cystic
spaces containing mucinous material(arrow).
|
Histologically, there is
perivascular pseudorosettes and true ependymal rosettes. These are seen
as rosette like arrangements of cells with ependymal differentiation.
Electron microscopy reveals these vascular elements surrounded by an
acellular zone. Presence of pleomorphism, increased cell density, and
mitosis suggest anaplasia.
There are numerous variants including
common cellular, papillary and clear cell and the uncommon tanycytic, a
fibrillar variant. They are not of clinical relevance.
Ependymomas are characteristically GFAP
positive by immunohistochemistry.
Familial cases have been identified, but
the cause of most cases remains unknown. Cytogenic analysis has
characterized the loss of chromosome 22q as the most common genetic
abnormality. As more than 50% of them have lost or altered chromosome 22q
sequences, the possibility exists that a tumor suppressor gene important
in ependymomas is located on this chromosome.
Clinical presentation:
Intraventricular ones present with
features of hydrocephalus.
Cranial neuropathy can occur in 25% of cases, due to compression or
invasion of the floor of the fourth ventricle. Spinal seeding is
suspected in the presence of backache and radicular symptoms.
Hemispheric ones usually with seizures
and neurological deficit.
Spinal cord tumors present with signs
and symptoms of an intramedullary
lesion.
Imaging:
Radiologically, the CT reveals an iso to
hypodense enhancing peri/intra ventricular lesion with different degrees
of calcifications, necrotic and cystic changes. It is iso to hypodense on
T1 and hyperdense on T2 MRI images. Hemorrhage is reported in 10% of
cases. A tumor-vermis cleavage plane in a posterior fossa tumor that is
isodense on CT is highly suggestive of ependymoma.
Management:
Surgical excision and post
operative irrradiation have been the mainstay of treatment.
Uncontrolled studies suggest that total surgical
resection offers long term remission. Second stage surgery, if
needed, has been recommended to to achieve total excision. Recent
advances in surgical tools, intraoperative imaging, and
electrophysiological monitoring have greatly helped the surgeon in his
goal of total excision.
Regardless of location, craniospinal
radiation is advocated both for cases with evidence of spinal
seeding and for high grade ones. In the absence of spinal seeding, some
consider spinal radiation less useful.
Most surgeons add cervical radiation in
all infratentorial cases, although not supported by controlled studies.
Ependymomas
are traditionally considered to be one of the most radiosensitive brain
tumors. Furthermore, low grade ependymomas represent the most
radioresponsive tumors within the glioma group. Although the need
for postoperative high-dose radiation therapy to achieve local tumor
control is generally agreed upon, there remains a controversy as to the
extent of irradiation. Recent studies of postoperative radiation
therapy did not reveal an improvement in survival by additional
prophylactic spinal irradiation. Thus, local control remains the main
therapeutic challenge in the treatment of intracranial ependymomas.
Recommended
doses are 50-60Gy for the primary tumor, 45-60Gy for whole brain
irradiation and 30-40Gy for the spine. Children should receive 80%
of adult doses. A few study groups use interstitial irradiation for the
treatment of ependymomas.
However,
the role of radiotherapy in low grade cases where total tumor resection
has been achieved remains unanswered.
Ependymomas
have been reported to be relatively less responsive to chemotherapy.
Responses have been documented to nitrosoureas and vincristine.
However, despite a variety of protocols, improvement of progression free
interval by chemotherapy in addition to surgery and radiotherapy has not
yet been established.
Currently,
the indication for adjuvant chemotherapy is mostly restricted to
recurrences of anaplastic ependymomas in childhood.
Use
of stem cells to enhance the effectiveness of chemotherapy is being
studied.
Genetherapy and immunotherapy are under
trial.
Prognosis:
The prognosis has improved recently.
With complete excision, 5 year survival rates of 37% to 69%.
The prognosis is poorer in the very
young, in recurrences and in anaplastic variants. The main cause of death
in ependymomas patients is recurrence at the primary tumor site. Median
survival time after diagnosis of spinal seeding is 6 months.
Infratentorial and anaplastic
ependymomas metastasize most frequently.
Nearly two thirds of spinal metastases
previously reported originated from an anaplastic infratentorial
ependymoma.
According to autopsy data, spinal
seeding can be expected in 25% of cases subsequent to surgery of the
primary tumor.
Extraneural metastasis is rare; they are
usually from the supratentorial ones, and carry worse prognosis..
SUBEPENDYMOMA:
They are also called subependymal giant
cell astrocytoma (SEGA).
These are thought to be derived from
glial elements of the subependymal tissues found just beneath the
ependymal epithelial tissues found just beneath the ependymal epithelial
layer. They are slow growing, intraventricular tumors whose diagnosis is
most commonly made in adults, particularly in older men.
Familial
occurrences has been described. Subependymomas are most often
asymptomatic and incidentally found at autopsy.
They can be supratentorial (27%),
infratentorial (71%),or cervicothoracic(2%). Lateral ventricles is the
most common site, with obstruction at foramen munro and resultant
hydrocephalus.
|
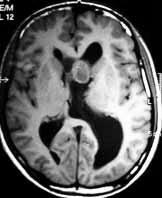
CT reveals an
heterogeneous iso to hypodense intraventricular mass, with variable
enhancement. The heterogeneity is due to cystic and calcific changes.
It is hypo to isodense on T1, and hyperdense on T2 MRI images.
Unlike ependymomas, there is no
transventricular extension.
Grossly, they are lobulated,
well circumscribed, nonencapsulated mass, with an intraventricularly
directed growth pattern that is expansile rather than infiltrative.
Histologically, they
resemble normal subependymal structures. Both ependymal and astrocytes
are seen.
Immunohistochemistry reveals
GFAP positivity.
The presence of ependymal cells
suggest an aggressive nature, and GFAP staining may be negative. These
SGAs may not be associated with Tuberous sclerosis.
Management includes
observation of incidental ones.
Those causing hydrocephalus
need to be excised through a transcortical or transcallosal
approach; the aim is to reestablish the CSF drainage; persistent
hydrocephalus may need a shunt procedure. Presently, there is no role
for radiotherapy or chemotherapy.
The patients with Tuberous
sclerosis may have associated with cardiac abnormalities, and hence
require a pre operative assessment.
Outcome is favorable, so long
as the hydrocephalus is adequately managed.
|
|
|
|
SEGA- CT
|
|
|
|
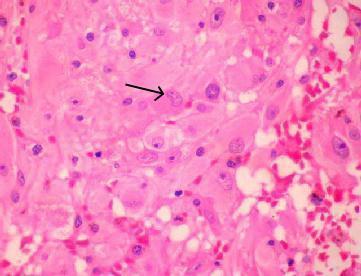
SEGA (H&E)- typical
cellular heterogenicity with large pyramidal like giant cells(arrow)
are admixed with spindle shaped and smaller fibrillated astrocytes.
|
|
|
