|
The term 'Primitive
neuroectodermal tumors (PNET)' may be used as a more descriptive term
to encompass all forms of embryonal cell tumor, with additional
references made to histology. It was originally intended to describe a
smaller subset of highly undifferentiated neoplasms, also of neural tube
origin. The majority of these embryonal tumors are found infratentorially
in the form of cerebellar medulloblastomas.
Recently, WHO opted
against this, retaining it instead for the purpose of referring to
cerebellar medulloblastomas, irrespective of location. Medulloblastomas are discussed in
this review. Other embryonal tumors
are discussed elsewhere.
Epidemiology:
They
comprise 6% of all intracranial neoplasms, about 12 per cent of all
neuroectodermal tumors and about 26 per cent of intracranial tumors in
children. While the majority of medulloblastomas are encountered
between the ages of 4 and 14 years, the first decade of life accounting
for more than 50 per cent, they do occur in infants and in young adults.
There
is a slight male preponderance. Though a number of authorities consider
the tumor to be restricted to the pediatric age group, according to Rubinstein
et al most a third of them occur between the ages of 15 and 35
years.
Pathology:
Medulloblastoma
is associated with several syndromes, and phakomatoses. 5%
of Gorlin's syndrome (multiple nevoid basal cell carcinomas,
multiple skeletal and cutaneous anomalies, calcification of the dura,
hydrocephalus, and developmental delay) develop desmoplastic medullblastoma.
It is an autosomal dominant disorder in which the defective gene is at
chromosome 9q. In Turcot's syndrome (mutiple familial polyposis),
the inheritance of the medulloblastoma is variable either autosomal
recessive or dominant, with defective gene on chromosome 5q21. Other
associated syndrome include, Li-Fraumeni syndrome, and
Ataxia-telangiectasia.
However,
no specific genetic abnormality or growth factor has been consistently
associated with its pathogenesis.
In
50% of them the chromosome 17p is absent. An isochromosome of the long
arm of 17q is thought to be related to tumor progression. The hsNF5
gene on chromosome 22 has been found to be altered in sporadic
medulloblastomas and PNETs (atypical teratoid -rhabdoid tumors of the
cerebellum).
The
medulloblastoma is an almost exclusively cerebellar tumor, made up of
primitive, poorly differentiated cells. While most investigators, concede
at least a neuroepithelial origin to the medulloblastoma, some believe
that there is no such tumor entity, it being merely an intermediate stage
in the development of an astrocytoma, an oligodendroglioma or an
ependymoma.
It is generally
conceded that there is no such cell as the medulloblast, and the tumor
possibly arises from the fetal external granular layer of the cerebellar
cortex, which normally persists till the age of one year.
In older
children and adults, the tumor may possibly originate from the inner
granular layer.
Alternate
cells of origin of the cerbellar medulloblastoma are the persisting cell nests
in the posterior or anterior medullary velum.
Thus
the medulloblastoma might arise from any of these germinative cell
groups, anywhere along their migratory path. This would determine
the location of these tumors in terms of a midline or a more lateral
situation, the former occurring at an earlier age, as the migration of
these primitive cells from the roof of the fourth ventricle would occur
sooner to the vermian cortex than to the more distant lateral cortex.
Hemispheric involvement is more frequent in adults.
The
gross appearance of the tumor is of some neurosurgical and practical
importance. The more cellular midline tumors of childhood often
appear circumscribed, are white, soft and friable, while the hemispheric
tumors of adult age are firm or even had to feel, darker and often
plaque-like over the cerebellar cortical surface. The former may be
found extending into the fourth ventricle and create doubts about an
ependymoma. The latter may present as a surface tumor simulating a
meningioma or as a mass in the cerebellopontine angle mimicking a
schwannoma.
|
Histologically,
the
'classic' variety consists of closely packed
fairly
uniform, small, undifferentiated cells in no particular arrangement, or
may be made up of the typical carrot shaped cells bearing central
eosinophilic cores (the Homer-Wright rosettes). Perivascular
pseudo-rosettes, of the type seen in ependymomas, may be
encountered in parts, in a small
proportion
of medulloblastomas. Mitotic figures may or may not be
encountered in medulloblastomas, and giant cell formation is distinctly
rare. Foci of necrosis and a high mitotic count are
usually
present.
Neuronal
differentiation (rosette formation and a small number of ganglionic
cells), astrocyte differentiation (GFAP immunolabelling of some tumor
cells), and the exceptional occurrence of
myoblasts
or melanin all reflect the large range of protein expression that
medulloblastomas can exhibit.
|
|
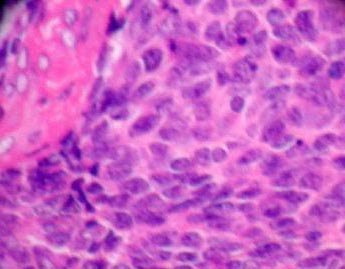
|
|
Medulloblastoma
(H&E):
closely packed uniform
small,
undifferentiated cells.
|
|
A
frequent histopathologic change in medulloblastomas is their tendency to
develop a rich reticulin frame work when they reach the surface of the
cerebellum, constituting a truly ‘desmoblastic’
change(20%). This may closely resemble the entity, described as
arachnoid sarcomas of the posterior fossa. On contacting the highly
vascular piaarachnoid, the medulloblastoma develops a moderate to profuse
mass of reticulin fibrils among the tumor cells. The desmoblastic
tumors occur in patients with a significantly higher mean age (21 years)
and are in the cerebellar hemisphere.
Electron
microscopic examination of medulloblastomas reveals large closely apposed
cells with occasional desmosomes pointing to the primitive nature of the
cells. While microtubules were detected within these as in other
primitive neuroepithelial tumors, synaptic structures have not been
encountered.
Metastasis,
generally arising by seeding of the parent tumor in the neuraxis is well
known, with 25% incidence at autopsy.
2%
to 7% metastasize extracranially, the commonest site being the bone.
Other sites include, the lymphatic spaces, peritoneum, lungs, and liver.
Clinical
Features:
Features
of raised intracranial pressure is the presenting symptom. There is an
higher frequency of hydrocephalus in children due to higher incidence if
vermian involvement. Unsteady gait, ataxia, and decreased coordination
are other features. rarely, there may be an head tilt (due to diplopia
related to sixth nerve palsy) or torticollis (secondary to pain because
of dural traction).
|
Backache
and radicular pain may indicate spinal seedings.
Imaging:
On
CT, it is an hyperdense, homogenously enhancing mass, with cystic or
necrotic areas. It is hypo to isodense on T1 and hyper to isodense on
T2 MRI images, with varying degree of gadolinium enhancement.
Associated hydrocephalus is seen.
Differential
diagnosis include, ependymoma, astrocytoma, metastasis,
hemangioblastoma, and choid plexus papilloma. Lateral lesions may mimic
schwannoma and meningioma.
Initial
evaluation should include, preoperative staging with neuraxis imaging,
and CSF analysis in possible cases.
|
|
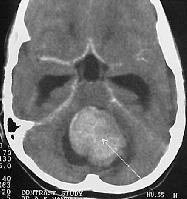
|
|
Medulloblastoma- MRI (axial)
|
|
|
Staging:
Whenever
possible, imaging of neuroaxis and CSF cytology should be done
preoperatively. MRI at 24-48 hours post operatively to determine the
residual tumor and CSF analysis 2-3 weeks post operatively should be
included in the study.
Negative
cytology does not rule out spread.
The
majority has no metastasis at presentation.
The
Chang system with various modifications, is used widely for
multicenter clinical trials.
Management:
|
|
Tumor (T) stage
|
Metastasis (M) stage
|
|
T1: <3cm in
diameter, involving one posterior fossa structure.
T2: <3cm in
diameter, invading 2 or more posterior structures.
T3a: >3cm in
diameter, invading
2 or more posterior fossa
structures.
T3b: Tumor
invading the floor of 4th ventricle.
T4: Tumor
extending out of the
4th ventricle, upward into
the
third ventricle, caudally
into the cisterna magna, or associated
with severe hydrocephalus.
|
Mo: No
evidence of tumor dissemination.
M1: Positive
lumbar CSF cytology.
M2:
Intracranial tumor dissemination.
M3:
Intraspinal dissemination.
M4: Systemic
dissemination.
|
|
Standard
therapy for medulloblastoma is surgical resection, followed by
craniospinal radiation. Supratentorial PNETs are staged and treated the
same way.
Radical
resection is
possible in the majority. The aim is to establish diagnosis, and restore
CSF pathways. Although the spread of tumor cells along the shunt appears
to be unfounded, current trend is to avoid a prior shunt procedure for
the common (75%) associated hydrocephalus. An extraventricular drainage
at the time of tumor resection helps.
A
transvermian approach, through a vertical incision over the lower
vermis, is widely practiced.
Cerebellomedullary
fissure approach avoids vermian incision. This approach involves opening
the fissure between the tonsil and medulla, the PICA (posterior inferior
cerebellar artery) is in close proximity. The fissure is widened by
lifting the tonsil off the medulla, the choroid plexus is visualized with
the tumor anterior to it. The fissure is widened on both sides for
a better exposure.
Transforamen
magendie approach is ideal for large tumors (65%), which grow through
the foramen and occupy the space between tonsils and the cisterna magna.
Meticulous
debulking of the tumor followed by dissection from the surroundings is
carried out. The part of the tumor covering the brainstem (30% of the
tumors are adherent to the brainstem), should be removed last, because
the large space created by tumor excision enhances the visibility.
Incidence
of post operative new neurological deficits have been reported to be in
the range of 28% to 44%. Abducent and facial nerve palsy, worsening of
cerebellar dysfunction, bulbar palsy, brainstem injury are the
possibilities. These are labeled as the 'fourth ventricle syndrome'. In
the majority, the deficits do not recover. Post operative seizures can
occur in about 7% of cases.
'Cerebellar
mutism'
following posterior fossa surgery was initially described by Hirsh
in 1979, and occurs in 5% to 30% of cases, usually in children.
Classically, it occurs on 1 to 2 post op days. There is irritability,
decreased verbal output, and behavioral disturbances, global cerebellar
dysfunction, and weakness of limbs. Cranial neuropathies can occur. This
is self limiting and recovery takes 4 days to 4 months. In majority the
recovery is not complete and require long term supportive care.
The
damage to the dentate nucleus and the dentate-thalamo-cortical connection
seems to be the cause, as it happens more commonly after excision of
large vermian lesions, necessitating incision and retraction of the
dentate nucleus. Patients with brainstem tumor involvement are at a
higher risk. SPECT studies reveal hypoperfusion of the thalamus, medial
frontal lobe, and the frontotemporal-parietal regions in the acute phase.
Standard
treatment includes radiotherapy. 5 year survival rates of 50% to
70% with 5400 to 5800cGy administered to the posterior fossa and 3500cGy
to the neuroaxis have been reported. Attempts, such as
hyperfractionated irradiation and dose reduction, have been made to
decrease cognitive deficits associated with craniospinal irradiation in
patients younger than 3 years old; they resulted in increase in relapses.
More recent attempts include the use of chemotherapy with encouraging
results. Focal radiation and stereotactic radiosurgery have
been shown to be acceptable adjuvants to conventional irradiation.
Additionally,
chemotherapy in patients with incomplete resection, or with metastasis,
improves the 5 year survival rates to 90%, and in those with metastasis
to 50%. Various chemotherapy regimens in association with radiotherapy
have been tried. Recently, it has been reported that, vincristine weekly
during radiotherapy and then vincristine, cisplatin, and CCNU for eight
cycles afterwards, substantially improves survival. New agents that have
shown promise in laboratory models of these tumors, are being studied in
patients. In addition, the use of blood stem cells to permit high dose
chemotherapy that would otherwise cause dangerously low blood counts is
being explored. Shorter, more intensive chemotherapy is currently being
tested in children. Studies are on to see the effectiveness of
chemotherapy to delay the radiotherapy in the young children.
To
summarize, the widely recommended regimen include,
1) Surgery and craniospinal radiation in those above 3 years old.
Chemotherapy is added in those with residual tumor and in those with
metastasis.
2) Surgery in those under 3 years old; radiotherapy is delayed until 3
years old.
Preirradiation chemotherapy may be considered.
Outcome:
Age
at diagnosis and presence of spread decides the survival.
Patients under 3 years of age belong to poor risk.
Duration
of symptoms, severity of hydrocephalus, tumor size, and even brainstem
invasion do not correlate with survival.
The
extent of tumor resection does influence the survival significantly,
especially in standard risk patients (the patients older than 3
years with no evidence of metastasis). Those with residual tumor volume
of less than 1.5cm2 have shown improved 5year progression free
survival in 77%, compared with 53% in those with more than 1.5cm2
of residual tumor.
None
of the biological indicators, such as GFAP, DNA ploidy, have been shown
to correlate with outcome consistently.
Treatment
sequelae
include, cognitive and neuropsychological dysfunction, endocrinopathy,
and secondary malignancy. Progressive decline in overall intelligence is
noted in almost all children, especially under 7 years old, receiving
craniospinal radiation. It is most evident 2-3 years after treatment.
Many long term survivors suffer psychological difficulties in their adult
years. Growth retardation, thyroid dysfunction, delayed puberty, and
adrenocortical insufficiency may also occur.
Second
malignancies, have occurred 6-7 years after initial therapy, and 50% are
in the radiotherapy field. Acute leukemia has been reported in children
treated with craniospinal radiation and chemotherapy.
Recurrence
is
usually incurable. Treatment of recurrence include resurgery and a
variety of chemotherapeutic agents. Use of stem cells and even bone
marrow transplantation have been tried with some success.
Those
with asymptomatic recurrences survived longer than those with symptomatic
recurrences. The majority of recurrences present within the first two
years. Aggressive surveillance with brain and spinal MRI is recommended.
Those who had no recurrence after 8 years, can be considered cured.
|
