|
Phakomatoses
is the coined by van der Hoeve in 1920, to describe a group of
hereditary neurological disorders that
have cutaneous and ocular stigmata. It is derived fro Greek root 'Phako'
meaning birth mark/mother spot. They are a group of disorders
characterized as dysplasias or neoplasms of organs derived from the
embryonic ectoderm. They have common features of neuroepidermal
maldevelopment and undifferentiated cells with disturbed patterns of cell
migration.
They
are commonly divided into:
1)
Neurofibromatosis, 2) Tuberous sclerosis, 3)von Hippel-Lindau disease,
and 4) Neurocutaneous angiomatosis.
All
phakomatoses do not manifest ocular and cutaneous
findings;
VHL
shows no skin markers and the neurocutaneous
angiomatoses no ocular lesions.
Many
similarities exist between these groups, and several families have been
reported with overlapping manifestations of two phakomatoses. The genetic
abnormality in many of these disorders has not yet been identified and the fact that stigmata of
more than one of these syndromes have been
seen in the same patient could indicate that they are all
due to an abnormality in a small group of genes.
The
recognition of one of these syndromes in a patient mandates genetic
screening of other family members to provide genetic counseling. Finally, the CNS abnormalities can exist with out these disorders.
NEUROFIBROMATOSIS:
This is the commonest of the phakomatoses, with a reported incidence of one in 3000 births. It is an
autosomal dominant disease with high penetrance, but variable expression.
This syndrome has been variously classified in literature, but recently
consensus is for two types which account for over 95% of all cases. Other cases of neurofibromatosis
represent either poorly expressed or variant
types.
|
Neurofilbromatosis-l
(NF-1):
NF-1 previously termed von Recklinghausen neurofibromatosis or peripheral neurofibromatosis, was
first described by von Recklinghausen,
in 1882. The genetic abnormality is
thought to be in chromosome 17 and is of extremely variable expression,
with members of the same family showing marked differences in clinical features.
A
diagnosis of NF-1 is made, if the patient fulfills any two of the following criteria:
a) Two or more neurofibromas of any type or
one plexiform neurofibroma
b) Six or more cafe-au-lait skin macules visible in room light,
each 5 mm or more in size in
prepubertal patients; or,
15mm or more in post pubertal patients.
c) Two or more Lisch nodules.
d) Optic glioma.
e) Axillary or inguinal freckling.
f) Characteristic osseous lesions such
as sphenoid dysplasia or thinning of long bone cortices with or without
pseudoarthosis.
g) A first degree relative (parent, sibling or offspring) by the above
criteria.
Not all
the patients of NF-1 fulfill the criteria given above. These patients must be presumed to have
the NF-1 gene, but with poor gene expression.
1)
Cutaneous neurofibromas are characteristic of NF-1. These Schwann cell
tumors occur on the distal cutaneous nerve endings. They are most
numerous in the thoraco abdominal region, and the presence of
neurofibromas on the nipple or areola of the breast suggests an
association with pigmentation and / or hormones. They
do not pose any serious problem to the patient except cosmetic, or rarely pain or itching.
Operative removal of the lesion is done for
painful or irritant lesions or
for cosmetic purposes.
2)
Plexiform neurofibromas may form along
the course of any nerve. While they grow mostly
from distal sensory nerves, they tend with growth to engulf major nerve
trunks and motor branches, rendering operative removal difficult with
attendant risks of a major neuro deficit. If it is asymptomatic, it is
best alone.
There is
a definite risk of malignant transformation in these patients. Neurofibrosarcoma
occurs in about five per cent of patients and is
the most dreaded complication of this disease. Treatment involves
amputation of the limb and major
resection, followed by radiotherapy and chemotherapy. However, 5 year survival rates
are only around 23%.
3) Lisch nodules arc pigmented hamartomas of the iris. They are present
in upto 94% of NF1 patients and seen, usually , after puberty.
4)Though spinal neurofibromas occur mostly
on the dorsal nerve root, the ventral roots may also he involved. These are often multiple and are most common in the cervical and lumbar regions. Surgery is necessary if cord compression develops.
The rare
occurrence of neurofibromas within the spinal cord, is seen more often
in case of neurofibromatosis than in the general population. Other spinal cord tumors
are not a prominent feature in NF-1.
5) Optic nerve gliomas occur in about
5-10 % of patients with NF-1. The
tumors behave like hamartomas and
the treatment is as for these tumors occurring in the general population. Brainstem and
post.fossa are other common sites. Hydrocephalus due to other tumors
and those due to aquedect stenosis are more common in NF-1.
6)
Macrocephaly, learning disorders, sphenoidal wing dysplasias,
peudoarthosis, pheochromocytoma and kyphoscoliosis are the other
lesions in NF1.
|
|
|
|
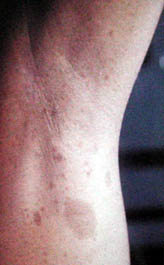
Axillary
freckling with
large
cafe-au-lait spots
|
|
|
|
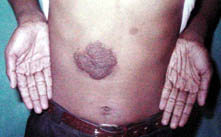
Giant cafe-au-lait spots
|
|
|
|
NF1with Rt.optic glioma
|
|
|
|
Plexiform
neurofibroma
with
palmar freckling
|
|
Neurofibromatosis-2
(NF-2):
This was previously called central neurofibromatosis or bilateral acoustic neurofibromatosis. It is an autosomal dominant disease, with the genetic abnormality on chromosome 22. Though the method of gene expression is not clear. It is much less common than NF-1.
|
The
criteria for the diagnosis of NF-2 are
a) Radiological evidence of bilateral acoustic neuromas
or
b)
a first degree relative with NF-2 and either a unilateral acoustic neuroma, or
two of the following: Neuro
fibroma, schwannoma,
meningioma, glioma, juvenile
posterior subcapsular cataract.
Patients
with NF-2 present with bilateral acoustic neuromas, which are for the
majority, symmetrical and present
with symptoms during adolescence and early adulthood.
|
|
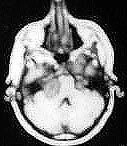
|
|
Bil.Acoustic neuromas-MRI
|
|
|
A diagnosis of NF-2 should be suspected in any patient below
30 years of age, who has an acoustic neuroma, in a patient with multiple
meningiomas and in patients with Schwann cell tumors and
minimal stigmata of NF-1. All such patients and family members of NF-2 patients
should be screened for bilateral acoustic tumors with BAER,
contrast enhanced high resolution CT and/or MR.
Patients
with NF-2 are liable to have other tumors including multiple Schwann cell tumors on
peripheral nerves, spinal roots and cranial
nerves, cranial and spinal astrocytomas
and meningiomas. Treatment of these patients
is aimed at maintaining brainstem and spinal cord function. Surgery is offered for the larger
tumors first, while small tumors without any
major pressure effects are kept under observation.
|
|
|
|
Small cafe-au-lait spots
|
|
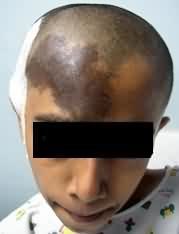
TUBEROSE SCLEROSIS (Bourneville’s disease or Epiloia):
Von
Recklinghausen, in 1862, described association
between cardiac myomas and brain sclerosis.
Bournville, in 1880, correlated cortical tubers, seizure and mental
retardation, and called it ' tuberous sclerosis'.
Vogt
Henrich, in 1908, described the clinical triad- adenoma
sebaceum, seizures, and mental retardation.
|
Tuberose sclerosis (TS) is an autosomal dominant
disorder with a variable penetrance. The
incidence is about 1 in 10,000 births and the extent of expression is
very variable. More than 60% are new mutation (i.e. no family history).
The
gene responsible is thought to be on chromosome 9q34 and 16p13.
Skin
manifestations:
Ash
leaf spots and other depigmented macules
are,
best seen under a Wood's lamp
(ultraviolet light).
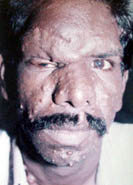
Adenoma
sebaceum is an angiofibroma, a progressive lesion which develops after
birth and shows rapid growth around puberty. It has a characteristic distribution, over the cheeks, nose, and
chin, sparing the upper lip and often confused for acne vulgaris.
Shagreen
or sharkskin patches are dermal fibromas which usually develop after 10
years of age. They occur mostly in
the lumbosacral region. They are not pathognomonic of TS and may occur
in isolation.
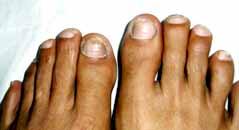
Ungual
fibromas or Koenen's tumors are angiofibromas which occur in the lateral nail
groove,
along the proximal nail fold or under
the nail. They are more common in the toes than in the fingers.
Nervous system manifestations:
Cortical plaques (or tubers) and
subependymal glial nodules are
developmental hamartomas containing glial and
neuronal cell populations,
which do not enlarge once
brain growth has stopped. There
is no evidence of malignant transformation
in these lesions. Degenerative changes take place with gliosis and umbilication of the
cortex at the site of the tubers, leaving normal
brain in between.
Subependymal nodules (SEN), <1 cm
in size, are scattered along the entire wall of the lateral and third
ventricles. They are mainly glial hamartomas and contain calcium
deposits.
Subependymal 'giant cell astrocytomas' (SEGA) occur in about
10% of the patients with TS and do not develop from the subependymai nodules. These tumors react negatively to GFAP and are probably
neuronal in origin. They probably arise from the
germinal cell matrix which explains their vascularity. Anaplastic transformation is rare. Calcified
or hypodense lesions may also be seen in the
posterior fossa. They may be GFAP positive or
negative, but S-100and NSE positive.
No
isolated case of SEGA, without TS, has been reported.
|
|
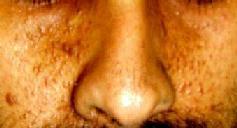
|
|
Adenoma
sebaceum
|
|
|
|
Ungual fibromas
|
|
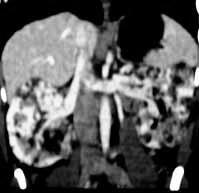
|
|
Bil.Renal angiofibromas-MRI
|
|
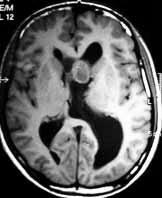
|
|
SEGA-MRI
|
|
Lesions are also seen in other organs such as the
retina, heart, kidney and lungs.
In the eye, retinal hamartomas occur.
They rarely lead to visual problems.
Cardiac rhabdomyomas occur
in about 30% of patients; they may remain asymptomatic or may produce
heart failure in infancy. Renal angiomyolipomas are commonly found in these
patients.
Clinical features:
The clinical diagnosis of TS can be made by Vogt's
triad of seizures, mental deficit and
adenoma sebaceum.
The
disorder can, however, present variably.
In
fetal stage, ultrasound or fetal echocardiogram may reveal hydrocephalus,
and cardiac myoma.
At
birth, ash leaf spots and infantile spasms are characteristic.
In children, seizures predominate. The seizures are
mostly tonic-clonic or infantile myoclonic, though partial motor and complex partial seizures are also
seen. Petit mal attacks are lot common. The degree of mental retardation in these patients varies and regression
has been noticed in older patients. 45% of them may have normal intelligence.
Severely
affected children exhibit bizarre purposeless hand movements and posture,
but no true athetosis or chorea.
In later life,
there may be spastic hemiplegia or diplegia. They
may be due to either uncontrolled seizures or to the development of a
brain tumor. Motor deficits are rare, though they may be seen as a
manifestation of a brain tumor. There may be features of obstructive
hydrocephalus, due to intraventricular SEGA.
Roach et al has listed the following criteria for the diagnosis of TS.
|
Primary features
|
Secondary features.
|
Tertiary features.
|
|
Facial angiofibromas.
|
Affected 1st degree
relative.
|
Hypomelonoic nodules.
|
|
Multiple ungual fibromas.
|
Cardiac rhabdomyoma
(HPE/scan +ve).
|
'Confetti" skin
lesions.
|
|
Cortical tubers (HPE +ve).
|
Cortical tubers (CT+ve).
|
Renal cysts.
|
|
Subependymal nodules or
subependymal giant astrocstrocytoma (SEGA).
|
Noncalcified subependymal
nodules (CT+ve).
|
Enamel pits.
|
|
Multiple calcified subependymal
nodules protruding into ventricles (CT+ve).
|
Shagreen patches.
|
Hamartomatous rectal polyps
(HPE+ve).
|
|
Multiple retinal
hamartomas.
|
Forehead plaque.
|
Bone cyst (CT +ve).
|
|
|
Pulmonary
Lymphangiomyomatosis (HPE+ve).
|
Pulmonary
lymphangiomyomatosis (CT+ve).
|
|
|
Renal Angiomyolipoma
(HPE+ve).
|
White matter heterotopias
(CT+ve).
|
|
|
Renal cysts (HPE+ve).
|
Hamartomas of other organs
(HPE+ve).
|
|
|
|
Infantile spasms.
|
Definite: one primary and
two sec or one sec and two tertiary;
Probable: one sec and one
tertiary or three tertiary;
Suspected: one sec or two
tertiary.
Imaging:
On CT,
cortical tubers, unless calcified, are difficult
to identify. The calcifications are mostly just
lateral to the foramen of Munro and in the body of
the lateral ventricle, though rarely, they may occur in
the wall of the third and fourth ventricles.The tendency for these lesions to protrude
into the lateral ventricle,
distinguishes them from other calcified lesions seen in cytomegalovirus
infection, cysticercosis and toxoplasmosis. Other
lesions which may encroach
into the ventricles, e.g. heterotopic grey matter or ependymomas are not
multiply and do not calcify.
Venticulomegaly, perhaps due to high CSF protein may be seen in children.
MRI
scan of the brain is the most
sensitive study. Cortical tubers appear as focally expanded gyri, which
do not enhance. They are iso/hypodense intracerebral
lesions are seen especially in the frontal, parietal and occipital lobes,
represent areas of defective myelination and heterotopic hamartomatous
tissue which occur particularly at the junction of grey and white matter.
These are seen in 12-69% of cases, but are not diagnostic of TS when they
occur in isolation.
Bands of abnormal
signal intensity radiating from the ventricles to the cortical mantle and
/ or wedge shaped lesions with their apex at the ventricle and their base at a cortical tuber. White matter
mass lesions appear less often; they are hyperdense on T2 and
hypo/isodense on T1.
Subependymal (SEGA) tumors are seen as isodense lesions enhancing
uniformly with contrast. They are located mainly at the foramen of Munro and produce obstructive hydrocephalus. They may, occasionally,
have a cystic component.
Asymmetrical ventricular dilatation may be seen in the absence of tumor.
Management:
Treatment involves controlling seizures with
antiepileptic drugs and special education for
the mentally handicapped.
ACTH ,
and lesionectomy is highly selected patients may help. The
life expectancy of these patients is decreased,
the causes of death being cardiac failure, brain tumors, status epilepticus and renal
failure.
No need for
surgical intervention in asymptomatic patients. Asymptromatic
children must be followed up periodically.
The
brain tumors can be excised in adults with a good prognosis;
subtotal excision to relieve hydrocephalus,may suffice.
In children, the associated
hydrocephalus may be shunted, and the child is reviewed periodically.
Role of radiotherapy is controversial.
VON
HIPPEL-LINDAU DISEASE (retino cerebellar angiomatosis):
The
von Hippel-Lindau (VHL) disease or complex is an autosomal dominant disorder with variable
expression, characterized by either more than one hemangioblastoma within the neuraxis associated with at least one
visceral manifestation. No cutaneous stigmata
arc seen in patients with VHL complex. It has an incidence of about one
in 40,000 live births. It is probably caused by a gene complex that maps
to the short arm of chromosome 3.
The
association of retinal, cerebellar and visceral
lesions was made, in 1926. by Arvid Lindau, who started his work
by investigating cerebellar cysts. The retinal angiomas had been described earlier, by Collins, in
1894 and by von Hippel, in 1904. Brandt
published the autopsy results of von Hippel's patient
and described tumors in the viscera in addition to
those in the brain and the spinal cord.
Retinal angiomas arc seen in over 50
per cent of patients with the VHL complex and may be the only finding in
children under 10 years of age. The lesions are seen mostly in the peripheral parts of the retina, though they
have also been recorded at the macula and
the optic disc. They are usually seen
in both the eyes.. Photocoagulation is the treatment of choice. As new lesions may appear
in course of time, the patients must be kept under regular ophthalmological follow up.
|
The typical lesion in
the neuraxis is a cerebellar hemangioblastoma, which at autopsy is found in at
least 60% of patients with the disease. Hemangioblastomas may also be found in the brain stem, spinal cord
and in the supratentorial compartment. The lesions may be solid or cystic.
They are commonly multiple, with the tumors appearing
metachronously.
Angiomas may also be found in other
organs such as the liver, spleen, kidney, lung, the skeletal system,
epididymis, and the renal cortex.
However, the most common and
dangerous tumors are pheochromocytoma and renal cell carcinoma, which cause death in a significant proportion of VHL patients. Renal cell
carcinoma is seen in up to 25 per cent of patients with the VHL complex, and differs from its sporadic
counterpart in its earlier age of onset, multicentricity, and
synchronous or metachronous bilateral involvement.
|
|
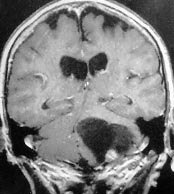
|
|
Cystic
hemangioblastoma
-MRI
(cor)
|
|
NEUROCUTANEOUS ANGIOMATOSES:
These are a group of genetic
disorders which have an abnormality
of blood vessels of the skin and nervous system as their only common feature, and arc grouped together for
convenience. Each syndrome has other
systemic angiomata as well as hematopoietic and immunological
deficiencies.
Ataxia
telangiectasia (Louis-Bar or Border-Sedgwick syndrome) is an
autosomal recessive disorder with progressive ataxia, cutaneous
telangiectasias. Prognosis is poor, with death usually occurring in the
second decade due to infection or neoplasia as a
|
result
of humoral and cellular immunodeficiency.
Sturge-Weber syndrome (Encephlotrigeminal angiomatosis) may be caused by a somatic
mutation occurring sporadically, rather than as an inherited disorder.
The characteristic skin lesion is a unilateral facial angioma (pot-wine
stain) in one or two dermatomes of the trigeminal nerve. There is an
ipsilateral parieto-occipital leptomeningeal venous angiomatosis with
underlying cortical atrophy. Calcification of the second and third
cortical layers of this region appear as ' rail road' calcification on
plain x-rays of the skull. Patient presents with seizures or
hemiparesis. SAH is rare.
In Klippel-Trennauney-Weber
syndrome (spinal cutaneous angiomatosis), the cutaneous angioma is
unilateral on the body, involving one or more
|
|
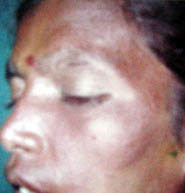
|
|
Port wine stain
|
|
dermatomes, with a
hemangioma of the spinal cord at the same level.
The lesion is seen
as a spinal variant of Sturge weber syndrome.
Fabry's
disease (Angiokeratoma Corporis Diffusum) results from the
accumulation of ceramide trihexoside in the media and endothelium of
small blood vessels, due to a deficiency of alpha galactosidase. It is an
X linked recessive disorder, characterized by telangiectasias of the
lower half of the body. Skin lesions apart, renal function may be
impaired with resultant hypertension and myocardial infarction. More
severe forms have diffuse involvement of vessels of the peripheral nerves
and of the CNS, leading to CVAs in young adults. Painful polyneuropathy
is another neurological problem.
Other rarer
syndromes include
Fibrous
dysplasia (Albright's syndrome)-dysplasia of bones,
irregular cafe-au-lait pigmentation, sexual precocity in females,
endocrine disturbances, mental retardation, seizures and 'ground glass'
radilogic appearance of bones.
Osler-weber-Rendu
syndrome (Hereditary hemorrhagic telengiectasia)-autosomal
dominant disease with angiomas of the skin, mucosal surfaces, and nervous
system, and usually presents with hemorrhage and should be excluded in
all patients with multiple AVMs, or family history of SAH, or repeated
epistaxis.
Wyburn-Mason
syndrome -AVM in the midbrain with unilateral retinal and
facial malformations.
Neurocutaneous
melanosis (Rokitansky-van Bogaert syndrome)-cutaneous
by pigmented nevi, intracerebral melanotic pigmentations, and
hydrocephalus.
Incontinentia
pigmenti (Bloch-Sulzberger syndrome)-cutaneous bullae,
verrucation, crustation and pigmentations, and cerebral palsy and
seizures. Ocular, skeletal, and cerebral malformations may be there.
Multiple
nevoid basal cell carcinoma (Ward-Gorin-Goltz
syndrome)-multiple basal cell carcinomas. skeletal anomalies,
congenital hydrocephalus, medulloblastoma, visceral cysts and
malformations.
Cutaneomeningospinal
angiomatosis (Berenbruch-Cushing-Cobb syndrome)-cutaneous
vascular nevus, angiomas in spinal cord, vertebrae and viscera.
Systemic
angiomatosis (Ullmann's syndrome)-cavernous and telangiectatic
angiomatosis of CNS and viscera, cutaneous angioma.
Oculocerebral
angiomatosis (Bregeats's syndrome)-oculo-orbital angiomatosis,
thalamoencephalic angioma, cutaneous angioma.
Neurocutaneous
lipomatosis-intracranial and intraspinal lipomata,
leptomeningeal lipomatosis, facial; and axial cutaneous lipomata,
visceral lipomatosis, skull lipomatosis, cranial, cerebral, and spinal
cord anomalies.
|
