|
Head injury remains a serious cause of mortality and
morbidity. From the moment of the trauma, a cascade of events takes place
and several different pathological events are set in motion. These events
are poorly understood. However, these events interact and determine the
final outcome. The patients' age and associated medical factors do play
an important role.
PATHOPHYSIOLOGY:
Axonal damage:
This occurs in various degrees in all head injuries due to
movement of the brain within the cranium as seen in
acceleration-deceleration injuries. Additionally, there are changes at
the subcellular level.
|
Diffuse axonal
injury is thought to
be responsible for prolonged coma in patients without a mass lesion. In
increasing severity the damage may be graded as follows:
grade 1- damage in the white matter of the
hemispheres,
the
corpus callosum, the brainstem, and the cerebellum.
grade 2-also a focal lesion in the corpus callosum.
|
|
|
|
|
|
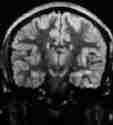
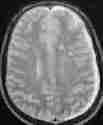
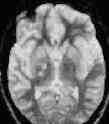
medial temp.lobe
|
corpus callosum
|
thalamus
|
|
|
grade 2 axonal injury, hyperdense
lesions-MRI T2
|
|
grade 3- in addition a focal lesion in the dorsolateral part
of the rostral brainstem.
The damage extend centripetally from the cortex to the
brainstem as the injury force increases; the direction of the
acceleration is also important. Sagittal acceleration only occasionally produces grade 1
damage. Coronal acceleration (lateral movement) has a high
incidence of diffuse axonal damage. Oblique acceleration causes
intermediate type. A higher grade is related deeper and more prolonged
coma as well as more severe residual neurological deficit.
At first there is progressive accumulation of organelles
associated with axonal thickening and later large ball- or club- like
swellings. Ultimately, they lose contact with the distal segment
resulting in axonal disruption. Myelin sheath breaks down followed by
phagocytic cell reaction.
Histological post-mortem studies reveal
axonal retraction balls in short (days) survivors, large numbers of
microglial stars formed by reactive astrocytes in intermediate (weeks)
survivors and signs of demyelination of the tracts in the longest
(months) survivors.
Vascular damage:
Structural cellular changes, and changes in autoregulation
occur following injury.
Endothelial lesions in the pial arterioles appear in the
form of a baloon or bleb; they burst into the lumen and crater-like
lesions are formed. Abnormal permeability of the vascular wall to
macromolecules with perivascular leakage. There is dilatation of
arterioles immediately after injury. There is a decrease in
vasoconstrictor response to hypocapnia and in severe injury the ability
to react is lost. Hyperaemic responses to arterial hypoxia are reduced as
well. These changes impair CBF.
Brain ischaemia:
Mass lesions, arterial injuries, vasospasm, increased ICP,
loss of autoregulation, and brain swelling can interact resulting in
ischaemia. A CBF below the threshold for infarction (18ml/100gms/minute)
creates ischaemia with loss of neuronal activity. The extent of damage
depends on severity and duration of ischaemia. After decompression of a
mass, there is sudden reperfusion and there a risk of increased ICP
and edema in some, especially after a long period of compression as often
seen in delayed decompressive surgery.
Brain
edema:
The post traumatic edema is predominately vasogenic due to
ischaemia; cytotoxic edema may compound the problem. There is blood brain
barrier breakdown and leakage of plasma constituents extracellularly. In addition,
cell swelling also occurs as a result of inadequate Na+ K+ pumps and
influx of Na+ into the cells.
Increased
intracranial pressure:
This may be a result of various processes and in turn may aggravate
different processes. The volume of the mass lesion, changes in CSF
absorption, changes in CSF volume, compliance of the brain tissue, and
the cerebrovascular volume and pressure determine the ICP.
Brain herniation:
Rapidly expanding lesions soon block CSF spaces causing
pressure gradients in the parenchyma. Eventually, there is a descent and
herniation of the medial temporal lobe in the supratentorial lesions and
cerebellar tonsils in the infratentorial lesions. Herniation causes
interruption of CSF passage and distortion of the brainstem and
compressive ischaemia. This results in further raise in ICP and
deterioration of the patient.
Biochemical changes:
Following head injury, neurochemical processes evolve
gradually in time and lead to structural damage. A detailed understanding
is still awaited.
There is an increase in extracellular K+ and release
of neurotransmitters, especially the the excitatory aminoacids (EAA) such
as glutamate; this, in turn, leads to further massive increases in
the k+ efflux and the Ca++ influx. Ca++ can activate proteolysis that
will breakdown the cytoskeleton. Phospholipase C activity is
markedly increased; there is release of arachidonate from tissue
phospholipid sources. The arachidonate gets converted into the endoperoxide
prostaglandin G2 (PGG2) by cyclooxygenase. PGG2 gets converted to
prostaglandin H2 (PGH2) by prostaglandin hydroperoxidase (PGH); in the
presence of suitable cosubstrates such as NADH or NADPH; oxygen radical superoxide
is produced in the wall of cerebral vessels. Superoxide can react with
hydrogen peroxide (H2O2) in the presence of iron and form the extremely
reactive hydroxyl radical (OH) which can react with almost every
molecule found in living cells.
Oxygen radicals can mediate the vascular consequences and
vasogenic brain edema after injury. Other effects are lipid peroxidation,
release of Ca++ stores, inhibition of enzymes, mitochondrial destruction,
DNA damage and disruption of cytoskeleton.
MEDICAL MANAGEMENT:
On arrival, and initial resuscitation,
basic bedside investigations are done, including ECG, chest x-ray. An
ultra sound scanning of the abdomen to rule out a silent visceral
injury is advisable. Appropriate x-rays to rule out a bony injury and
x-rays of the cervical spines should be carried out in all patients, even
in those with no obvious cervical injury.
|
CT scan of the brain is the imaging of choice in all head
injuries. Skull x-rays have become redundant these days.
MRI of the spines is indicated when there is a suspicion
of a spinal injury.
Further management of head trauma is aimed
to prevent secondary insults to the brain in addition to attempting to
provide optimum conditions for recovery from the primary injury,
providing adequate cerebral blood flow and oxygenation.
|
|
|
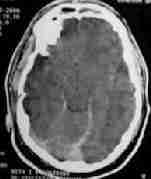
|
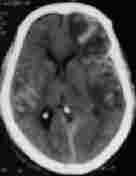
|
|
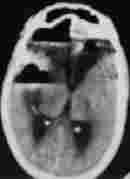
Pneumocephalus
|
Traumatic SAH
|
bil. hgic. contusions
|
|
|
Some of the traumatic lesions which require intensive medical care.
|
|
Following initial assessment and investigations, the
patients may be grouped into four groups for the purpose of planning
further care:
Group 1:
Patients with transient loss of consciousness, but are alert
without neurological deficit at presentation.
The neurosurgeon must exercise clinical judgment. Age and
systemic illnesses and alcohol intoxication may be considered. It is
safer to admit them for observation, as there is 1-3% risk of delayed
deterioration despite a normal CT.
Group 2:
Patients with impaired consciousness or a focal neurological
deficit; they may follow a simple command. GCS score may be between
12-9.The initial CT brain may be normal.
They require admission for strict observation.
ICP monitoring may be considered if they have a non-surgical
lesion in the CT scan, especially if they require urgent surgical
intervention for an associated injury.
It is prudent to repeat the CT after a day in this group.
Group 3:
Patients who are unable to follow even a simple command and
those with a GCS score of less than 8 fall in this group.
Immediate intubation and controlled positive pressure
ventilation may be considered. CT scan and other investigations should be
carried out immediately and appropriate measures taken.
Anti edema measures may be considered in the waiting period.
Those who do not require surgical intervention must be
ventilated.
Careful monitoring of the patient’s status is the foundation
of intensive care.
Lately, bedside measurement of CBF, metabolism, and electrical
activity has become a routine in some centers. Serial clinical
examination is still the most comprehensive method of assessing the
progress of the patient.
a) Respiratory care:
Neurogenic pulmonary edema is being
increasingly recognized lately in severe head injury. Hypothalamic injury
resulting in sympathetic discharge, systemic hypertension, left sided
heart failure and pulmonary hypertension and edema is blamed. In addition
there appears to be a centrally mediated effect on alveolar capillary
endothelium that increases its permeability.
Hypoxia is common even in those with no obvious cause for
the decreased ventilation.This has been termed ‘central neurogenic
pulmonary insufficiency’. Post-concussion apnea resulting in military
atelectasis, incipient neurogenic pulmonary edema, and pharyngeal
dysfunction with aspiration are the possible causes.
Associated thoracic injury, and
aspiration are other possible causes. Fat embolism, more often seen with
long bone fractures, occurs in other disease states as well. DVT and
pulmonary embolism are to be kept in mind.
Adult respiratory distress syndrome may
develop as a result of the above conditions, compounding the problem.
Removal of any inciting factors and ventilatory
care are the mainstay.
It is a good practice to obtain ABG analysis immediately on
arrival as the pulse-oxymeter may be deceptive and administer
supplemental oxygen to the injured.
The decision to ventilate the patient depends on whether the
patient is :
a) conscious or unconscious with no other significant
injury.
b) able to maintain adequate airway with good cough
reflex, and without stridor or not.
c) able to maintain optimal oxygenation (spO2>95%
and pO2 of 100-120mmHg and pCO2 of 28-33 mmHg, but< 45 mmHg) on his
own or not.
d) normotensive or not.
Patients with stridor require an airway, oro/naso
pharyngeal.
If the pO2 is <60mmHg and /or pCO2 >40mmHg, and in
those with GCS of <8 (irrespective of ABG parameters), endotracheal
tube and ventilation is preferred.
If the patient requires to be ventilated, slow rates and
large tidal volumes (12 breaths a minute and 10ml/kg of tidal volume) are
recommended to ensure adequate venous drainage and re-expand atelectatic
areas. PO2 must be maintained between 100-140 mmHg as higher pO2
induces cerebral vasoconstriction PCO2 must be maintained between
28-35mmHg and lesser pCO2 will compromise CBF. It is important to
maintain the cerebral perfusion pressure (CPP= MAP - ICP)) of 70-100 mm
Hg. Concommitent use of ICP monitor and arterial pressure monitor is
essential for the same.
Dehydration due to osmotherapy causes metabolic acidosis
which may stimulate respiration in under sedated patients on a
ventilator and reduce the pCO2 to less than desired to normalise
the pH, which will compromise the CBF. Metabolic acidosis is the earliest
sign of dehydration before it shows up in serum urea and creatinine
levels. Dehydration must be avoided.
Mini heparin and antiembolic stockings help to prevent DVT.
b) Cardiovascular care:
Severe head injury induces induces hyperadrenergic state
resulting in hyperdynamic cardiovascular and metabolic response and
arrythmias and signs of myocardial ischemia
Cerebrovascular autoregulation is frequently deranged in
association and may interact with CVS abnormalities to affect CBF.
Hypotension leads to cerebral ischemia. Systemic hypertension may lead to
cerebral hyperemia and swelling.
Meticulous attention to CVS function is thus important.
Sustained systolic hypertension above 160mmHg must be treated.
Propranolal is an appropriate agent. Sodium nitroprusside, because of its
cerebrovascular dilatory effect, causes cerebral swelling. Some advise
fluid restriction. But, it is preferable to maintain a state of
normovolemia with isotonic crystalloid or colloidal solutions.
c) Fluid and electrolytes care:
Both pathophysiological processes and therapeutic maneuvers
may lead to a number of electrolyte
abnormalities. Sodium is the most important one. Serum
electrolytes and osmolality should be checked frequently and attended to
appropriately.
d) Hematological care:
Anemia reduces the oxygen carrying capacity of
the blood; however, the oxygen carrying capacity also depends blood flow,
which increases with a fall in blood viscosity (hematocrit). In fact, the
oxygen delivery increases until an hematocrit of about 33%. Hematocrit
below 30% should be corrected with blood transfusions.
Coagulopathy due to release of brain thromboplastin and
damage to cerebrovascular endothelium is often seen. Disorders range from
abnormalities in lab findings to full blown disseminated intravascular
coagulation.
Measurement of hemoglobin, thromboplastin time, partial
thromboplastin time, platelet count, fibrinogen levels, and fibrinogen
degradation products are widely used.
Treatment is not satisfactory; obviously the underlying
cause should be eliminated. Blood loss should be corrected along with
fresh frozen plasma and platelets. Low dose heparin may help.
e) Gastrointestinal care:
Problems of gastritis to frank ulceration are associated
with head injuries, especially in the first week. About 10% of them have
significant bleeding. Pathogenesis is not clear. Vagal stimulation due to
diencephalic or brainstem injury, adrenergic surge have all been
blamed.
Cimitidine (500mg I.V) is effective; concurrent antacids
help.
f) Seizures:
About 5% of the head injured patients have seizures in the
immediate post injury period. Debate continues on prophylactic anti
convulsants; most surgeons use them in severe head injury.
Acute control
and status epilepticus are attended to appropriately.
g) Infections:
Meningitis
results from open or penetrating wounds, CSF fistulae, ICP
monitoring, and operative procedures. Meningitis complicating basilar
fractures within 72 hours of injury is commonly due to streptococcus
pneumoniae. Meningitis from open wounds or CSF leaks or following
craniotomy often results from staphylococcus aureus or gram negative
bacillus or both. Staphylococcus aureus and epidermidis are the common organisms
in meningitis following ICP monitoring.
Strict aseptic precautions during procedures, and use of
appropriate antibiotics help.
h) Nutrition:
Starved head injured patients lose sufficient N2 to lose
weight by 15% per week.
Major head injury alone may provoke a hypermetabolic state
as seen in multiple trauma and burns. Concurrent injuries, fever, sepsis,
seizures, and posturing may further aggravate.
The aim is to optimize the nutritional support
until homeostasis is re-established as the illness subsides.
Overfeeding is to be avoided. For periods longer than few days, the
help of a qualified dietician is mandatory.
i) Increased ICP control:
It is widely accepted that increased ICP
is a cause and an effect of brain injury. There are conditions in
which patients have severe neurological deficit with out increased ICP.
In the absence of a mass lesion, increased ICP is seen only in 30% of diffuse
brain injuries.
It is the main concern of medical
management.
j) Cerebral protection:
Since secondary damage seems to be due
to a cascade of of biochemical reactions, there are multiple
possibilities of pharmacological intervention. Various clinical trials
are initiated and conducted on the brain protective effect in the head
injured of several different pharmacological agents.
Nimodipine in high doses (60mgm every 4
hour) is reported to have beneficial effect on injured brain. Some
studies lately have found no benefit. NMDA and AMPA antagonists are
being tried to counteract glutamate excitotoxicity on experimental basis.
Free radical scavengers such as steroids, mannitol, barbiturates, vitamin
C and E have been used in various combinations.
Hypothermia reduces CMR, edema
formation, release of glutamate, excitatory transmitters, ion exchange,
blood coagulation, and the concentration of lactate and leukotrienes. It
is reduced in some centers with good results.
Group 4:
The patients in this group show no brainstem activity (brain death).
The neurosurgeon’s role becomes a social one, comforting the
family.
|
