|
A dysraphic lesion is one that
relates to the midline closure of the neural tube, which is complete in
the human embryo by the 25th day of intrauterine development. It may be ,
more commonly spinal or cranial. Cranial dysraphisms
are discussed elsewhere.
Embryology and
Pathogenesis and antenatal diagnosis are discussed elsewhere.
The
term ‘Spinal dysraphism’ covers a range of developmental abnormalities of
the spinal cord and its surroundings. It includes both conditions obvious
at birth or before (spina bifida aperta) and consists of, a vertebral
defect is associated with a cystic mass on the back and conditions that
are not obvious at birth (spina bifida occulta), but consists of a defect
in the vertebral arches not associated with an externally visible sac on
the back.
Spina
bifida represents a spectrum of conditions of disordered development of
the spine and spinal cord. In its most severe form open myelomeningocele.
The neurological deficits can be profound. Spina bifida may be open
(aperta) or closed (occulta)
Spina bifida aperta (cystica):
There is an obvious defect in the dorasal midline of
the spine. Spina bifida aperta is further subdivided into the following
groups: (1) a meningocele without cord tissue within the sac; (2)
myelomeningocele in which spinal neural tissue forms part of the sac; and
(3) rachischisis, which is the most severe form and involves a widely
patent dorsal opening of the spine with or without residual cord tissue,
and is usually associated with anencephaly.
Seventy per cent of them are seen over the distal
thoracic, lumbar and sacral. Rarely, the lesion may be situated
anteriorly (thoracic or intra abdominal meningocoele) or anterolaterally.
Clinical features:
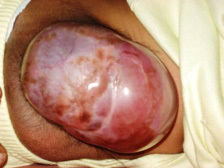
A meningocoele is sessile, sometimes
pedunculated and of variable size. It is covered by full-thickness skin,
which occasionally may be thin and translucent. It transilluminates
brilliantly with no evidence of neural tissue or strands. An impulse on
coughing or crying is apparent. There is no neurological deficit.
|
In a meningomyelocoele,
transillumination is positive and neural tissue may be demonstrable
within the sac. It is usually not reducible, though a crying impulse
and cross fluctuation with the open fontanelle may be elicited. The sac
is usually in communication with the subarachnoid space, but may also
be multiloculated and divided into non-communicating compartments by
fibrous or fibrolipomatous septa. The amount of neural tissue in the
sac varies from case to case. A few strands of ectopic nerve fibers to
the spinal cord or a part of the cauda equina may be found in the sac.
The neural tissue is closely adherent to the fundus with only flimsy
adhesions to the neck. Rarely such a meningomylocoele may be associated
with angiomatous, cartilaginous or lipomatous tissue.
|
|
|
|
meningocoele
|
|
In rachischisis (myelocoele, myelochisis),
the neural plate lies exposed on the surface as a reddish, vascular
granulating mass, usually lying in the middle and cranial part of the
defect. The lesion may be dry or if the central canal is patent, there
may be a continuous leak of CSF.
While meningocoeles present without any
neurological defect, a moderate to severe degree of deficit or even
complete loss of neuronal function is usual in cases of meningomyelocoele
or open myelocoele. The tethering effect of amyelomeningocoele can lead
to further deterioration as the child grows, even if the
myelomeningocoele is in the cervical region. The paralysis in lumbosacral
lesions is typically of the lower motor neuron type with flaccidity and
diminished or absent tendon reflexes. However, there may be evidence of
upper motor neuron involvement as well. In cervical and thoracic lesions
there is an upper motor neuron lesion with spasticity and exaggerated
jerks.
In children with spina bifida the
urinary tract may be affected in two ways. It is the site of a true
developmental malformation in about 20 per cent of cases but more
commonly there is a neurogenic dysfunction. Incontinence usually becomes
apparent after the age of one year. The important feature is regression
of already learnt bladder and bowel function. Absence of the anal reflex
and a lax anal sphincter indicate incontinence of the bowel.
Hydrocephalus is present in 80 per cent
of children with spina bifida aperta. The larger and more rostral the
lesion, the higher is the incidence. Studies in infants born with
meningomyelocoele and encephalocoele, showed that 96 per cent of the
paraplegic infants and 60 per cent of the non-paraplegic infants had
evidence of hydrocephalus. Imaging may reveal hydrocephalus in an
apparently normal-sized head. Hydrocephalus may become apparent only
after the excision of the meningomyelocoele. In the earlier years, this
was attributed to the removal of the myelocoele sac which was presurned
to act as an absorbing surface for the CSF. There has been no convincing
proof for this assumption. Aqueductal stenosis has been reported in 20-50
per cent of these cases. An associated Chiari malformation is probably
the commonest cause of hydrocephalus. Occasionaly, neurogenic laryngeal
stridor occurs in children with spina bifida and is most probably due to
the associated Arnold-Chiari malformation.
The incidence of associated congenital
anomalies elsewhere in the body is significantly higher in children with
spina bifida aperta than in the general population. Some of the
associated defects observed are congenital heart disease, dislocation of
hip, anomalies of vertebrae including hemivertebra and block vertebrae,
kyphoscoliosis, multiple rib defects, cleft lip and palate, ectopic
development of renal and intestinal tissue, and umbilical or inguinal
hernia. Developmental abnormalities of the urogenital tract are frequent.
Multiple meningocoeles or a combination of a spinal meningocoele with a
cranial encephalocoele is not uncommon.
Management:
Intrauterine repair of the
myelomeningocele is now possible, and early studies suggest that this may
decrease development of significant hydrocephalus without changing the
motor outcome, despite decreased exposure injury to the dysplastic cord.
Fetal therapy is a rapidly advancing specialty and an integral part of
interventional sonography. Open hysterotomy has been performed for the
repair of myelomeningocele, resection of sacrococcygeal teratoma in
fetuses with nonimmune hydrops, and treatment of an enlarging congenital
cystic adenomatoid malformation that is not amenable to thoracoamniotic
shunting. In-utero surgery to repair spinal defects is being tried. No
fetus has been cured, and published reports indicate no significant
improvement in the level of paralysis compared with optimal postnatal
care. However, approximately one third of the fetuses may show
improvement in Chiari malformation, decreasing the need for shunt
surgery.
Delivery of an infant with a suspected open
myelomeningocele should be by Cesarean section, in order to avoid the
risk of infection during passage along the birth canal; the child should
be nursed on its front or side, with a sterile moist dressing covering
the defect, and kept warm. Having examined the defect itself, clinical
assessment aims at determining the neurological deficit, both sensory and
motor. Much of this can be done by observation and gentle stimulation of
the limbs to ascertain sensation and movement. Bladder and bowel function
are difficult to assess with any certainty but a good urine stream may
suggest an incomplete deficit, although almost all children will go on to
have some degree of bladder and bowel disturbance.
Further examination is directed towards
possible associated congenital anomalies and hydrocephalus, as well as
general cardiopulmonary status. MRI may reveal associated lesions
such as intraspinal lipoma, dermoid or epidermoid, traction bands,
tethered and thickened filum terminale, a bony spicule or an ectopia of
the dural sac (occult intrasacral meningocoele), and also delineates a
Chiari malformation, syringomyelia and hydrocephalus.
The management of a pure meningocoele
involves a simple repair, while that of a meningomyelocoele or myelocoele
is prolonged, complicated and expensive. The treatment does not begin or
end with the surgical correction of the local defect, but begins from
the moment of birth and continues till such time as maximum possible
rehabilitation has been achieved. The total care of such a child requires
collaboration between the neurosurgeon, orthopedic surgeon, plastic
surgeon and the urologist. The help of a team of physiotherapists and
rehabilitation experts may also be necessary to give the child the
best-chance of self-sufficiency. Special educational facilities are
required as the child grows up.
Obviously, it is a problem in developing
countries.
Selective non-treatment, based on the
level of the lesion, the severity of associated hydrocephalus and degree
of spinal deformity, as well as the presence of other congenital
abnormalities, led to many severely affected infants not surviving. Not
all untreated infants succumb; they may go on to survive, with more
severe disabilities than had they been treated initially. Patients with extensive
paralysis, severe hydrocephalus, kyphosis and major associated congenital
anomalies in other systems be left unoperated.
Surgery for open myelomeningocele is
aimed at protecting the existing neural structures and preventing
infection. Surgery will not restore neurological function; nevertheless,
it is essential to preserve any functioning nervous tissue that does
exist. Because of the risk of infection, closure of the defect should be
carried out within 48 hours of birth. Closure of the defect involves
defining the neural placode and freeing this from arachnoid adhesions.
Some surgeons reconstitute the neural tube by folding over and suturing
the neural placode in an attempt to prevent future cord tethering;
however, this procedure is not essential and may unnecessarily damage
the delicate existing nervous tissue Care should be taken not to include
any skin appendage attached to the placode. The extradural space is
identified, the dura is mobilized and this plane developed around the
defect to allow closure of the dura in a watertight fashion. If
necessary, a dural graft may be tight fashion. If necessary, a dural
graft may be required to close the dura, without compromising the neural
structures and maintaining the closure free from tension. The muscle and
fascia on either side of the defect are mobilized; this may require
lateral releasing incisions if the defect is large, and then
approximated. The skin is then closed in a watertight manner. For very
large defects, plastic surgical procedures with myofascial or cutaneous
flaps may be required to achieve adequate closure.
Occasionally a pronounced kyphosis may
require surgical correction during the same procedure to make skin
closure easier and improve ventilation. Approximately 80% of children
with myelomeningocele will require a shunt at some stage. For those with
obviously severe hydrocephalus, this may need to be carried out within
several days of closure of the spinal defect. For those children less
severely affected, observation, with head circumference measurements and
assessment of signs of raised intra cranial pressure, will dictate the
need for and the timing of shunt insertion. The majority of children who
will need a shunt will do so by the age of 5 months.
Postoperatively, the child is nursed in
prone position to minimize adhesions of the cord to the dural suture
line, and close observation of anterior fontanelle and head
circumference. Hydrocephalus may require an external ventricular drainage
or ventriculoperitoneal shunt.
Associated deformities may require
specialised orthopaedic treatment. A careful urological evaluation and
institution of appropriate therapy to prevent ascending urinary infection
and hydronephrosis may be required. Physiotherapy and rehabilitation for
the residual neurological deficits have to be provided by the respective
specialists. The neurosurgeon's role continues through childhood.
Syringomyelia may develop and with the growth of the child, a tethering
lesion may manifest in about 20% at some point in their life.
Spina bifida occulta is discussed elsewhere.
|
