|
The worldwide
incidence of pineal tumor is about 1%. In Japan, Korea and China, it is
more common, constituting about 3 to 8% of all brain tumors, and about
10% of all pediatric brain tumors.
Pathology:
Histogenesis of
tumors of the pineal gland is related to the development of this
organ. In late fetal life as well as in early infancy, sections of
the pineal present a mosaic appearance produced by aggregation into
groups of large cells with large nuclei, with intervening bands of
smaller lymphocyte like cells with small dense nuclei. These are
immature forms of larger cells and disappear in childhood. In
adult life the gland is lobulated from the intersection of parenchymal
cells by connective tissue strands bearing blood vessels.
In the commonest
tumor of the pineal, now called ‘germinoma’ and considered as a
‘maldevelopmental tumor’, there is a recapitulation of the fetal
architecture of the pineal gland.
Most
pineal tumors originate infratentorially, and grow into posterior third
ventricle, and later into thalamus or posteriorly over the dorsal surface
of the quadrigeminal plate. Malignant tumors can invade the midbrain and
thalamus, which determines the resectability. The different cell types account
for the diverse pathology of pineal region tumors.
Pineal
tumors may be classifieds as follows.
|
A.
Tumors of pineal parenchymal cells (pinealomas) (about
25%)
1. Pineoblastomas
a. without differentition
b. with pineocytic differentiaition
c. with neuronal, glial or/and
retinoblastic differentiation
2. Pineocytomas
a. without differentiation
b. with normal differentiation only
c. with astrocytic differentiation
only
d. with divergent neuronal and
astrocytic differentiation (ganglioglioma)
|
B.
Germ cell tumors
1. Germinomas (about 70%)
2. Embryonal carcinoma, yolk sac tumor
and endodermal sinus tumors
3. Chorioncarcinomas
4. Teratomas
a. immature
b. mature
5. mixed germ cell neoplasm
C.
Tumors of glial cell origin
1. Astroctytoma
2. Glioblastoma
|
Other
rarer tumors include meningioma, ependymoma, paraganglioma, melanoma, hemangioma,
hemangiopericytoma, craniopharyngioma.
Rare
Cysts include
pineal cyst, arachnoid cyst,
epidermoid and dermoid, lipoma and metastases.
Pineal
Cyst:
These
benign pineal cysts are considered as normal variants of the pineal
gland. They consist of cystic structures surrounded by normal pineal
parenchyma. They are usually asymptomatic and incidental radiological
finding as a rim enhancing lesion compressing the gland. Very
occasionally, they may result in aquedect stenosis and require
intervention. Pineal cysts were visualized in 4.3% of normal patients in
one MR study as areas of high signal on intermediate T2-weighted images.
Pineal
parenchymal neoplasms (pinealomas):
They are rare,
constituting approximately 15% to 32% of all tumors in the pineal region.
Neoplasms of the pineal parenchymal cell are referred to as
pineoblastomas or pineocytomas depending on the cellularity and cytologic
characters of the tumors. Otherwise, these two sub varieties
are comparable in respect to the age of the patient and the gross
appearance of the tumor. Thus they both occur more frequently
in late childhood or early adulthood.
Pineocytoma is a better differentiated
variant of pineal tumors and occurs mostly in adult life.
Usually
well-circumscribed but may disseminate along the CSF pathways.
The
pineocytoma consists of small round cells set in a fibrillary background.
The cells may form Homer-Wright (neuroblastoma) rosettes, similar cells
are found in pinoeblastoma (the so-called 'ganglioglioma' of the pineal),
but the nuclear: cytoplasmic ratio is much higher in this tumor, and the
mitoses are evident.
Pineoblastoma
is
the least differentiated pineal parenchymal neoplasm. This tumor
tends to occur during the first or second decades of life. It is highly
malignant and represents a true primitive neuroectodermal tumor.
Usually replaces the tissue of pineal gland.
It
is pink, white or grey, smooth or granular when cut, sometimes cystic and
frequently hemorrhagic or necrotic.
It
bears a histological resemblance to medulloblastoma, the cells being
primitive with scanty cytoplasm and relatively large nuclei rich in
chromatin. An attempt at rosette formation may be seen.
The appearance may, in places, resemble a germinoma. The centre of the
rosette may not reveal argyrophilic filaments, which are more
readily seen in the rosettes of the pineocytoma.
On
account of their vascularity these tumors tend to bleed and the blood
pigment seeping out of the tumor may be detected in the CSF in the spinal
theca. Tumor cells may also be detected in the spinal fluid in
along the meninges.
Germ cell tumors:
Tumors
of glial cell
origin:
True gliomas of the pineal
gland are almost exclusively astrocytomas of varying grades of
malignancy. Being generally extensive, their precise source of
origin is difficult to determine and they might even originate from the
corpora quadrigemina. Small non-neoplastic glial cysts of the pineal may
be found incidentally at autopsy in the elderly.
Among the very rare pineal neoplasms may
be mentioned ganglioglioma and chemodectoma.
Clinical features:
Clinically, there are features of
hydrocephalus, such as, headache, vomiting, and papilledema.
Upward gaze paresis, convergence or
retraction nystagmus, and disturbed light reflex (Parinaud's syndrome)
is a characteristic sign due to midbrain compression at the level of
superior colliculus. Further compression, down gaze or horizontal gaze
can result.
Dorsal midbrain compression or
infiltration can cause lid retraction (Collier's sign) or ptosis.
Less commonly, 4th nerve palsies with
diplopia and head tilt can occur.
Ataxia and dysmetria can result due to
interference of superior cerebellar peduncles.
Rarely, there may be hearing
disturbances due to disturbance of inferior colliculi.
Endocrine disturbance is rare due to
tumor spread to hypothalamus.
Imaging:
CT and MRI typically reveals a
homogenously enhancing mass in most cases, depending on the pathology.
MRI delineates pineal region masses better than CT, showing the
relationships of the tumor to the posterior third ventricle, vein of
Galen, and aqueduct.
Pineal germinomas and primary pineal
tumors are most often isointense with the brain on T1- and T2-weighted
images. A few lesions exhibit long T1 and T2, which may correlate with
embryonal cell elements. These tumors are well defined and enhance to a
moderate degree, usually without central necrosis, cystic change, or
hemorrhage.
Meningiomas can appear very similar on
plain scan, but their intense enhancement may set them apart from other
lesions.
Gliomas infiltrate the tectum and
posterior walls of the third ventricle. They tend to be poorly
circumscribed. Edema is not a consistent finding, and enhancement is
variable. Larger gliomas in the splenium of the corpus callosum may
present as pineal region masses.
Teratomas are of mixed signal intensity,
frequently with calcification. They may also have cystic components and
fat.
Arachnoid cysts, epidermoid and dermoid
tumors can usually be distinguished from other pineal region tumors by
their increased signal on T2-weighted images.
|
|
|
|
|
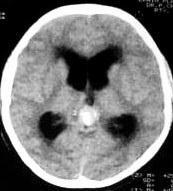
Pineal Germinoma-CT
|
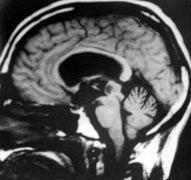
Pineoblastoma-MRI
|
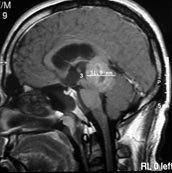
Pineal glioma-MRI
|
MR venography may be more useful to
study the displacement of deep venous system, which is very important in
deciding the surgical approach, is well shown in MRI. Meningiomas from
the velum interpositum and epidermoids or other tumors from the corpus
callosum displace the deep venous system ventrally and inferiorly which
may give a 'clue'.
Staging with neuroaxis imaging and if
possible, CSF analysis, is recommended.
Tumor markers:
CSF analysis, whenever possible, should
be carried out. It is most valuable in germ cell tumors, which constitutes
about
70% of the pineal tumors. Expression of
embryonal proteins, such as, alpha fetoprotein (AFB), and beta HCG are
indicative
of malignant germ cell elements. Other
biological markers for germ cell tumors include, Lactic dehydrogenase
isoenzymes
and placental Alkaline Phosphatase
(PLAP).
Pineal parenchymatous cell tumor markers
include, melatonin, and S antigen; they are more valuable in follow ups
and to study the response to treatment, and not reliable for diagnostic
purposes.
Management:
The pineal tumors are invariably,
aggressive. Gross total removal, although ideal, is technically
difficult. The main aim of management is to get a tissue diagnosis for
further therapy, and reestablish the CSF drainage pathway. The hydrocephalus is usually
managed with shunt surgery, although it carries a potential risk of tumor
dissemination.
In the past, the pineal tumors were
irradiated with out a tissue diagnosis, and surgery was performed in unresponsive
cases, as favored by the Japanese. With a good response, germinoma,
(which constitutes about 70% of pineal tumors, and found more frequently
in Japan) was presumed.
Nowadays, a tissue diagnosis is
recommended before radiotherapy, either by stereotactic biopsy
or open surgery.
Germinoma is an exception. Detection of
Placental Alkaline Phosphatase (PLAP) is diagnostic of germinomas, and
obviates biopsy.
Stereotactic biopsy, shunt for the
hydrocephalus, and CSF studies at the time of shunt surgery, followed by
radiotherapy in malignant lesions is widely accepted, despite the recent surgical tools and
microsurgical techniques.
The aim of surgery is to get a tissue
biopsy, and reestablish CSF drainage. Advocates of surgery claim, that
only open surgery can provide adequate tissue for a detailed biopsy
studies, and that surgery can provide a 'cure' in benign lesions. In
addition, with the recent advances in surgery, and anesthesia, the pineal
tumors, which was once a 'no go area' in the past, can be satisfactorily
excised. Successful tumor excision may obviate a shunt. An
extraventricular drainage at tumor surgery is increasingly practiced.
The surgical approaches can be
divided into, supratentorial, infratentorial and a combined supra and
infratentorial approach. Surgeon's familiarity largely decides the
approach, more than the tumor extensions.
Supratentorial approaches include,
parietal interhemispheric (as described by Dandy), and occipital
transtentorial approach (as described by Horrax, and later
modified by Poppen and others). It provides a wide exposure, and
is best suited for tumors extending supratentorially or laterally into
the trigone of the lateral ventricle; however, it is difficult to remove
the tumor that lies below the convergence of the deep venous system.
Infratentorial supracerebellar approach
was described by Krausse, and later popularized by Stein. It
is midline approach suitable for midline tumors that grow into the third
ventricle, and posterior fossa. The deep venous system caps the dorsal
aspects of the tumor, and remains away at surgery; however, the exposure
provides only a limited access to the lateral extensions of the
tumor, and the tumor above the deep venous system.
Complications: Pineal region
surgery is a microsurgical challenge, despite all new surgical tools. In
expert hands, severe permanent morbidity should be rare. Transient
worsening of extraocular problems, and gait, hemiparesis due to
brain stem retraction, visual field deficits due to occipital lobe lobe
retraction, may occur. Disconnection syndromes due to corpus callosum
incisions have been reported. Hemorrhage into the residual tumor bed is
the most devastating complication.
Germinomas are highly radiosenstive. A
long term survival of greater than 90% is reported with radiotherapy. The
recommended dose is 5500cGy given in 180cGy daily fractions, with 4000cGy
to the ventricular system, and an additional 1500cGy to the tumor bed.
Nongerminomas are less responsive with 5 years survival in about 40% of
cases.
Radiotherapy may be withheld following
total excision of benign lesions, such as pineocytoma; but they need
close follow ups. Combined radiotherapy and chemotherapy is being
studied to reduce the radiation dose in view of the reported delayed post
radiation complications. Stereotactic radiosurgery is
increasingly being used in pineal tumors with satisfying results; long
term follow up is awaited.
Staging with neuroaxis imaging and CSF
analysis, when possible, is recommended. Post operative craniospinal
radiation in tumors prone for dissemination is recommended.
Currently, prophylactic craniospinal radiation is not recommended.
Chemotherapy is, effective in children with
malignant pineal tumors. Many consider this as the first line of therapy
instead of radiotherapy in children under 3 years of age. It has a
definite role in recurrences. The agents include, carboplatin, etoposide,
and bleomycin, cyclophosphamide Vinblastine, losfamide in various
combinations and cycles.
|
